
结构生物化学/卷 3
结构生物化学在新型药物的开发中变得至关重要。现在,药物正使用生物化学工具(如X射线晶体学)进行研究。现代生物化学方法通常用于通过理解结构的折叠和弯曲来理解酶结构。酶是生物催化剂,通过降低形成反应过渡态所需的能量来提高反应速度。酶通常由蛋白质或蛋白质组组成。了解蛋白质的三级和四级结构可以告诉科学家药物是如何起作用的。药物科学家利用酶的结构,从旧药中开发出新药。药物通过首先让信息或药物接触细胞外部并使其接触受体来穿过细胞膜。然后,连接的换能器将信息传递到内部,最终被信号放大,促使细胞完成其功能。许多科学家认为,在 10 到 20 年内,医学领域将发生巨大变化,特别是在医生为患者开药的方式方面。目前,药物的给药是根据平均剂量进行的,这些剂量是根据个人的体型和年龄确定的。药物的有效性在称为“剂量-反应曲线”的图表中显示。创建这些图表是为了显示药物的预期效果与给药药物量之间的关系。通常,还有另一条曲线显示引起最大副作用的药物量。药理学家使用这些数据首先证明药物有效,然后用它为医生提供安全的药物剂量范围,以便为他们开药的患者使用。
好的药物设计依赖于许多变量,包括它在体内的吸收、它正确工作的活性、它将保持活性多长时间以及它的毒性。了解目标分子的结构可以更直接地找到适合目标形状的分子,从而创造出最有效的药物。因此,显然不适合目标的分子将自动被认为在其当前状态下不起作用,应被忽略或重新配置。[1]
未来,目标是为每个人提供量身定制的药物。这个想法是根据个人的DNA序列设计药物,该序列描述了个人独特的生物化学。背后的愿望是获得更有效且副作用更少的药物。仅仅在不到十年前,量身定制药物的愿望还只是一个不切实际的想法,但随着 DNA 测序技术的极大进步,这个梦想可能在几十年内成为现实。
纳米医学
[edit | edit source]每年被诊断患有癌症的人数仍然非常高。科学家一直在深入研究相对较新的纳米粒子药物设计领域,希望开发出更有效的抗癌药物。目前的化疗技术能有效地杀死癌细胞,但也对人体必需的健康细胞有毒。
纳米药物可以通过化学工程专门靶向癌细胞,而不会产生化疗的副作用。纳米粒子的不同部分可以被修饰,以便药物能够进入人体的血液并靶向癌细胞,而不会被肝脏分解。一些纳米粒子包装在脂质体中[check spelling],而许多新的化合物则通过生物降解聚合物传递。例如,聚乙二醇 (PEG) 是一种生物降解聚合物,可以保护纳米粒子不被免疫细胞识别,从而帮助药物到达目标位置。一些纳米粒子外壳由“环糊精”等糖类制成,但覆盖着 PEG。糖上的羟基使化合物可溶于水,但易在酸性环境中分解以释放药物。大约有 12 种基于纳米粒子的抗癌药物正在进行临床试验,等待批准在全球范围内分发。
纳米粒子还被用于将 RNA 分子传递到癌细胞,通过反义疗法靶向癌细胞。如果 RNA 分子在纳米粒子的帮助下能够到达癌细胞,那么它们就有可能与癌细胞自身的 RNA 结合并使某些基因失活。例如,反义疗法可以阻止癌细胞中蛋白质的产生,这有助于阻止癌症的整体生长。
用于药物的天然物质
[edit | edit source]天然产物以许多不同的方式帮助了医学。人们发现,天然产物含有许多抗病和抗癌特性。它们可以被合成到一种化合物中,用于药物。例如,像蓝细菌这样的小型植物状生物体存在于潮湿的环境中,它们具有强大的抗癌和杀菌来源。夏威夷大学马诺分校的迪克·摩尔教授能够想出一个方法来寻找对发展缓慢且难以治疗的肿瘤特别有效的化合物。一个例子是名为隐藻素-8 的化合物,它可以破坏各种用于小鼠的实体瘤的细胞支架。
此外,海洋中还有种类繁多的物质和化学物质具有极其强大的抗癌和抗病能力。这推动了科学家开发新的方法来合成地制造源于这些海洋天然产物的化合物。制造良药的目标是科学家对天然化合物进行研究,以提取它们的药用特性,但去除引起不良副作用的部分。

药物可以从最奇怪的材料中制成。例如,爱荷华大学的化学家吉姆·格洛尔一直在研究如何使用一种生活在动物粪便中的真菌来制造抗生素。这些生物体被称为腐生生物,这意味着它们喜欢粪便,在开发有用的药物方面具有巨大潜力。这些真菌会释放杀死附近物种的化学物质,这也是生物医学研究人员和科学家想要得到的,这样他们就可以开发出杀死对人类有害的不需要的真菌的药物。
疾病的生物化学
[edit | edit source]根据世界卫生组织的《国际疾病分类和相关健康问题》,目前对疾病的定义包括 22 个章节。这些章节被细分为 2500 多个区块,为我们提供了数千种疾病的表型描述符。
生物化学家专注于了解分子的结构以及这些分子结合在一起的过程。了解这些过程也让我们有机会在它们出错时进行“纠正”。但是,获得治疗方法需要考虑许多不同的关键因素。例如,在药物开发方面,它是化学、生物化学、药理学、毒理学等知识的结合。这显然不是一个可预测的过程,因为实验药物的失败率很高。
生物化学家研究疾病的生物化学,以研究他们可以用来开发新的疾病治疗方法的重要要点。该领域的主要要点包括非典型微生物代谢物的生物合成、基于结构的抑制剂设计、耐药机制以及蛋白质折叠动力学导致的蛋白质错误折叠和聚集的作用。
参考文献
[edit | edit source]Service, Robert F. “纳米粒子特洛伊木马从实验室奔向临床。”《科学》2010 年 10 月 15 日:314-315
戴维斯,艾莉森。“健康的化学”。《NIGMS》2006 年 8 月:36-42。http://publications.nigms.nih.gov/chemhealth/coh.pdf
在生物化学领域,为了治愈疾病,应该评估几个有助于药物设计的关键主题,包括基于结构的抑制剂设计、药物耐药机制、非典型微生物代谢产物的生物合成以及可能导致蛋白质错误折叠的蛋白质动力学作用。[1]
疾病的生物化学:迫切寻求天作之合《生物化学年度综述》第 78 卷:55-63(卷出版日期 2009 年 7 月)DOI:10.1146/annurev-biochem-120108-082254 约翰·W·科扎里奇 http://www.annualreviews.org/doi/abs/10.1146/annurev-biochem-120108-082254?journalCode=biochem
为了将药物传递到预期的目的地,靶向是药物递送中的一个重要主题。配体是一种与生物分子形成复合物以用于生物学目的的物质。有三种类型的配体通常用于药物递送靶向。抗体和/或抗体片段、肽和适体。根据靶向情况,使用不同类型的配体。
药物靶向可以定义为将药物递送至目标器官或系统的途径和效率。虽然这看起来可能是一个简单的过程(只需吃掉它或直接注射到血液中?),但实际上需要解决的障碍太多了,以至于制药公司需要花费数年时间才能开发出一种药物。这些挑战包括药物是否确实到达了目标器官,以及它是否以对人体有益的显著量到达了器官。众所周知,如果药物被吃掉,它最终会进入血液并到达身体的各个部位。通常人们不知道的是,药物是否在身体中分布得太稀薄而无用,以及这些药物到达非目标器官的后果。
药物浓度可能稀释到无任何效果的程度,这可能与制药公司试图找出这种药物的剂量有关。在测试药物时,科学家必须确定可以服用多少药物才能产生显著的效果,同时也要测试增加剂量是否会增加副作用。为了确定药物剂量,必须在这两者之间取得平衡。一些变量将包括患者的体重、年龄、血液水平、肾脏和肝脏的健康状况以及患者正在服用的其他药物。所有这些因素都将在确定向患者服用多少药物方面发挥作用。如果患者肝脏健康,很可能大部分药物会在到达血液之前被破坏。如果患者血量少,就不需要服用很多药物,因为它不会被稀释。患者服用的其他药物可能会发生化学反应,从而导致严重的副作用。另一个需要考虑的因素是药物的化学重量和性质。如果药物的分子量大,就不需要服用太多药物。
主要而言,制药公司更担心的是,原本用于肾脏的药物最终会出现在肺部附近,或者类似的事情。这被称为副作用。这通常通过观察细胞膜表面的碳水化合物链来解决,以发现特定器官的细胞具有哪些受体。开发一种适合大多数这些受体(一种广泛且结构通用的药物)的药物会增加副作用的可能性。因此,开发一种尽可能特异性地与细胞受体结合的药物以减少副作用至关重要。发现副作用并最大限度地减少这些副作用是使药物通过审查测试并进入药店货架的重要部分。(5)
两种在细胞膜水平上针对有害微生物的药物包括多粘菌素和制霉菌素。多粘菌素会干扰细菌的细胞膜,因此细菌无法作为渗透屏障发挥作用。多粘菌素和制霉菌素之间的功能区别在于制霉菌素会干扰真菌和酵母的细胞膜,而多粘菌素会破坏细菌的细胞膜。制霉菌素会与麦角固醇结合,麦角固醇是真菌细胞膜中的一种必需成分,这会导致细胞膜中断,表现为膜上出现孔洞。
氟喹诺酮类药物,或环丙沙星,通过阻止 DNA 复制所需酶(即 DNA 旋转酶)解压缩来抑制核酸(如 DNA 或 RNA)的合成。结果,没有 DNA 复制。氟喹诺酮类药物广谱且效力极强,因此可用于治疗难以治疗的细菌,例如炭疽杆菌(导致炭疽)和铜绿假单胞菌。另一种针对 DNA 水平的抗微生物药物是利福平,用于通过抑制原核生物 RNA 聚合酶来治疗结核病,这反过来会阻止转录,因此不会产生 mRNA。细菌没有这些必需的蛋白质就无法存活。
利奈唑胺,或先锋霉素,会破坏蛋白质合成的起始,因此用于治疗耐甲氧西林金黄色葡萄球菌 (MRSA) 和耐万古霉素肠球菌 (VRE),这两种病原体是最难治疗的病原体之一。其他作用于蛋白质合成水平的抗微生物药物包括链霉素、庆大霉素、四环素和红霉素。
“抗体片段”。抗体片段。N.p.,n.d. 网站。2012 年 10 月 28 日。<http://www.piercenet.com/browse.cfm?fldID=4E03B016-5056-8A76-4ECA-982DA6CAAC8A>。
“Creative Biolabs”。Creative Biolabs。N.p.,n.d. 网站。2012 年 10 月 28 日。<http://www.creative-biolabs.com/phagedisplay1.htm>。
Tortora,Gerard J.,Berdell R. Funke 和 Christine L. Case。微生物学:入门第 10 版。波士顿:本杰明·卡明斯 :, 2010 年。印刷版。| 第 20 章| 第 600 页}
5. 设计药物,美国卫生与公众服务部,NIH 出版物,2006 年 7 月转载
纳米医学是一个真正跨学科的领域,它涵盖了纳米工程、化学工程、生物工程、化学、材料科学、生物学、物理学、药学和医学等学科。为了确保药物在体内有效地发挥作用,必须充分考虑生物系统、药物的化学反应性和在体内的物理运输。纳米医学是近几十年来备受关注的研究领域。越来越多的创新方法使已知的药物更有效,例如将药物与两种或三种材料结合起来,以实现单一药物无法实现的目标。
量子点 - 适体缀合物示例 在癌症治疗中,重要的是要观察抗癌药物是杀死肿瘤组织还是正常健康组织。因此,成像是重要的应用。荧光和量子点是两个良好的指示器。量子点是一种类似于荧光的半导体纳米晶体,由于其独特的光学特性,包括宽吸收范围和窄光致发光光谱、高量子产率、低光漂白和抗化学降解性,在生物系统和标记中得到了越来越多的应用。量子点的表面可以修饰,使抗体、适体或肽键能够附着在其上。现在,这种复合物表现出多种对癌症治疗有用的特性。
这里给出的具体示例包括量子点、适体和多柔比星缀合物(如以下示意图所示),该缀合物靶向并杀死前列腺癌细胞。量子点荧光作为一种成像工具。RNA 适体的一端连接到量子点的表面,另一端连接到多柔比星。这种适体也起着活性配体的作用,靶向癌细胞。最后,多柔比星是一种众所周知的抗癌治疗剂,也具有微弱的荧光特性。多柔比星的荧光太弱,无法检测到;因此,它不可行。然而,如果使用量子点,就可以检测到薄的人体组织内的荧光信号。
当量子点单独存在时,它呈现绿色荧光。这被称为“开”状态。为了暂时“关闭”量子点的荧光,多柔比星的荧光特性在这个缀合物中变得至关重要。当两种材料都具有荧光特性并且彼此靠近时,它们会“猝灭”对方。在这种情况下,它们可以暂时禁用彼此的荧光特性。多柔比星呈现红色荧光,而这种特定的量子点呈现绿色荧光。当两者形成缀合物时,两种颜色能够“抵消”对方。抵消多柔比星与量子点之间的比例大约为 8:1。
在该缀合物进入癌细胞之前,它处于“关闭”状态。当缀合物通过对流和扩散在血流中运输到肿瘤部位时,由于缀合物上的适体配体,缀合物能够靶向肿瘤细胞。缀合物通过胞吞作用进入肿瘤细胞后,被送入溶酶体。溶酶体能够消化适体,分解缀合物。多柔比星的释放使量子点的荧光特性恢复到“开”状态,从而实现成像和检测。释放的多柔比星移动到细胞核,杀死癌细胞。以下图示是缀合物在癌细胞内的示意图。
这种创新的缀合物不仅能够靶向癌细胞,还能提供癌细胞内部的成像。还有许多其他方法有待发现,以实现更有效的靶向药物递送。
Bagalkot, Vaishali, Liangfang Zhang, Etgar Levy-Nissenbaum, Sangyong Jon, Philip W. Kantoff, Robert Langer, and Omid C. Farokhzad. "Quantum Dot−Aptamer Conjugates for Synchronous Cancer Imaging, Therapy, and Sensing of Drug Delivery Based on Bi-Fluorescence Resonance Energy Transfer."Nano Letters 7.10 (2007): 3065-070. Print. 纯药物通常与非活性物质混合以生产药物剂量(剂型)。这是出售给患者的药物的物理形式。在药学实践中广泛使用的常见剂型列示如下。
片剂通常以各种大小、颜色、形状和重量存在。它被称为药学实践中最流行的剂型,因为它具有紧凑、便携、准确、方便和无味的优点。在将混合物通过压片机进行机械压缩以生产药物的紧凑型固体剂量形式之前,将几种处方辅助剂(稀释剂、赋形剂、粘合剂、润滑剂、崩解剂、着色剂和调味剂)与活性成分混合。
然而,药物必须在胃中分解,以便以分子形式释放以发挥生物活性。这一过程有助于片剂形式的特性之一:作用开始缓慢。
一种可以咀嚼或在口中溶解后再吞服的片剂形式。
这种片剂不是为了在胃中溶解而设计的,而是为了在患者的肠道中分解。为了实现这一点,压片被涂上特定的物质,以防止它们在胃中融化。由于其特殊的结构,这些药物在吞服前禁止咀嚼或压碎。抗酸剂会导致在胃中溶解,因此禁止与肠溶片一起服用。
设计为放置在患者舌头下,以便活性成分可以立即被吸收进入血液,并在肝脏分解之前在体内首次循环。
放置在患者的牙龈和脸颊之间,因为这种药物将在那里缓慢溶解一段时间。
是使用水溶性材料包衣的药物,可以保护敏感药物免受光照和空气的劣化,掩盖药物的气味或味道,以便患者更容易服用。
其他名称是长效/延迟释放/延长作用片。这些片剂专门设计,以便活性成分以恒定速率缓慢代谢,持续时间较长,大约 8 到 24 小时。
呈卵形或圆盘形,以便缓慢溶解并长时间与口腔或咽喉接触。
被植入作为避孕或激素(如睾酮和雌二醇)的一种方法。这些圆柱形片剂被植入皮肤下方,以便药物能够缓慢地被吸收,持续很长时间。
是药物的固体形式,其中药物被包装在柔软或坚硬的明胶壳中。壳体大小从 000 号胶囊到 5 号胶囊(分别从最大到最小),并在胃中 10 到 30 分钟后溶解以释放药物。这种剂型的优点是它可以提供各种不同的形状和颜色,此外,还可以消除药物的味道和气味。
这种片剂利用碳酸氢钠与柠檬酸或酒石酸之间的酸碱反应,作为掩盖难闻苦味药物的掩味剂。 该反应将帮助物质溶解到溶液中,并通过释放二氧化碳气体引起“起泡”。
Reifman, Noah. 药剂师技术认证回顾。 第 9 版。 美国:AuthorHouse,2011 年。61-88。印刷。
是均质混合物,包含一种或多种可溶性成分(可以是液体、固体或气体),溶解在溶剂(水或与水混溶的液体)中。
这种剂型的优点是它可以在胃肠道中相对快速地吸收,更容易吞咽,因此通常用于儿童或老年人的剂量。 如果药物是外用溶液,则必须仔细贴标签。
这种类型的药物主要用于抗生素、抗组胺药、止咳药和维生素。 它被定义为甜的、粘稠的、浓缩的糖水溶液。
这可能是使用最广泛的液体剂型,因为它具有口感好、相对稳定且易于制备的特点。 它是甜的含酒精水溶液,酒精浓度可能会有所不同(不超过 20%)。
酊剂是另一种形式的酒精或含酒精水溶液,类似于酊剂,但酒精浓度更高。 植物、动物或化学物质是这种形式的主要成分。
包含细分散的不可溶性药物(称为内相),分散在水性外相中(通常包含额外的调味剂)。
患者可以通过口服或外用(如乳液)服用这些药物,或者根据分配给他们的悬浮液类型注射到体内。
可用于内服和外用制剂。 必须仔细阅读说明以了解乳剂是内用还是外用。 这些药物包含分散在油中的水(通过乳化剂稳定)或分散在水中的油(通过乳化剂稳定)以及微乳剂或透明乳剂。
Reifman, Noah. 药剂师技术认证回顾。 第 9 版。 美国:AuthorHouse,2011 年。61-88。印刷。
它是半固体制剂,具有多种功能,可以作为外用药物的载体、润肤剂(如润滑剂)或保护剂(防止皮肤受到刺激)。 软膏仅供外用,涂抹于皮肤或粘膜。
它们是类似软膏的制剂,比软膏更硬、更不油腻,吸水性更好。 由于这些特性,糊剂用于通过外用治疗渗出、湿性病变。
含有药物的半固体乳剂。 仅供外用。
细分散的固体药物,用于外用。 一些特定的粉末药物用于哮喘患者。
仅供外用制剂。 它由两个阶段组成:分散在粘性液体相中的固体内相。
通过粘附在患者的皮肤上,将药物以恒定、受控的剂量释放到血液中。 最常用的贴剂是硝酸甘油、东莨菪碱、尼古丁、雌激素和芬太尼贴剂。 警告:用酒精清洁。 反复多次应用于同一部位可能会引起刺激。
Reifman, Noah. 药剂师技术认证回顾。 第 9 版。 美国:AuthorHouse,2011 年。61–88。印刷。
它是无菌溶液,每次滴入眼睛 2 滴,药物与眼睛接触的时间非常短。 患者在滴入多种眼药水时,应在每次滴入之间等待 5 分钟。
眼膏用于滴入眼睛。 作为无菌乳剂,它们能够与眼睛保持更长时间的接触,而不是眼药水。
警告:使用后视力会模糊,因此患者应仅在睡前使用该药物。
在插入眼睛之前必须用药物预先浸泡; 它的优点是可以提供药物的控释和与眼睛更长时间的接触。
用于某些抗生素,如四环素和氯霉素。
药物放置在下眼睑与巩膜之间。其优势在于延长药物与眼睛的接触时间,控制释放到眼睛的药物量。使用前需要预先浸泡。
Reifman, Noah. 药剂师技术认证回顾。 第 9 版。 美国:AuthorHouse,2011 年。61-88。印刷。
它是一种固体剂型,在放置的地方会融化或溶解于水性分泌物中。它用于插入直肠、阴道或尿道,在那里它将发挥局部治疗作用或被患者的血液吸收。
成人和儿童栓剂的长度和重量不同。
- 成人直肠栓剂重约 2 克,长约 2.5 到 3.5 厘米,是儿童栓剂的大小和重量的两倍。
- 阴道栓剂呈球形、卵形或圆锥形,重 3 到 5 克。
- 尿道栓剂直径 3 到 5 毫米,长度在女性尿道和男性尿道中有所不同(女性 60 到 75,男性 100 到 150)。
这种剂型的药物以气体或空气的形式通过鼻子或嘴巴吸入,使微小的药物颗粒可以流入肺部的肺泡囊。
使用前需要充分摇匀。
大约 4 滴这种溶液或悬浮液可以滴入耳道。
警告:仅用于耳朵,如果药物为悬浮液,则必须充分摇匀。
它是通过球形注射器以室温插入直肠的液体制剂。这种药物的作用是局部的或全身的。
它是水溶液,用于引入患者体内的腔隙,主要目的是清洁腔隙。例如,眼部冲洗液用于去除眼睛中的异物;阴道冲洗液有助于清洁和提供阴道粘膜的药物,同时指向女性阴道。
Reifman, Noah. 药剂师技术认证回顾。 第 9 版。 美国:AuthorHouse,2011 年。61-88。印刷。
根据患者的症状,药物的不同给药方式是为了增强药物对身体的作用。下面是一个显示治疗窗的图示。研究人员的目标和挑战是最大限度地降低药物对身体的毒性,并最大限度地提高药物在体内的活性。为了使血液中药物浓度保持在治疗窗内,需要可持续的药物释放。为了实现这一点,许多纳米技术药物平台可用。最常见的是脂质体和水凝胶。
水凝胶可以将药物包裹在其核心。当环境发生变化时,水凝胶能够膨胀,从而导致药物释放。
pH 值的变化会导致膨胀,从而导致药物释放
离子强度的变化会导致凝胶内部离子浓度的变化,从而导致膨胀的变化,从而导致药物释放
电子给体化合物导致电荷和转移复合物的形成,导致膨胀发生变化,从而导致药物释放。
存在底物,酶促转化导致产物发生变化并膨胀,从而导致药物释放。
施加磁场以改变凝胶中的孔隙,从而改变膨胀,从而释放药物。
温度变化会导致聚合物-聚合物和水-聚合物相互作用发生变化,从而改变水凝胶的形状并导致药物释放。
施加电场会导致膜带电,并导致带电药物的电泳,并改变水凝胶的形状以释放药物
使用超声波照射来提高温度,导致水凝胶膨胀以释放药物。
张良方。 “控制药物递送系统”。 CENG 207 讲座 11。加州大学圣地亚哥分校,拉荷亚。2012 年 5 月 10 日。讲座。药物被归类为不同的主要治疗分类,描述了每种药物的治疗用途。
肾上腺的肾上腺皮质(位于肾脏附近)产生糖皮质激素,糖皮质激素在减少炎症反应中起着重要作用。这种类固醇有助于减少炎症迹象,如发红、肿胀、发热和炎症部位的压痛。治疗用途:糖皮质激素已被合成,广泛用于治疗药物血清和输血反应症状、支气管哮喘、过敏症,以及作为化疗的辅助药物。
1/ 麻醉性止痛药:止痛药通过与中枢神经系统内的阿片受体结合起作用。警告:嗜睡,与酒精一起使用时危险。在意外过量服用时,必须有麻醉拮抗剂可用。
2/ 非麻醉性止痛药和退热药: - 抑制前列腺素的合成。- 抑制其他使疼痛感受器敏感的物质。- 影响大脑的体温调节中心=> 退热。治疗用途:用于减轻疼痛和发烧。
抑制前列腺素的合成。治疗用途:产生抗炎、退热和止痛作用。谨慎:某些类型的这种药物是肠溶衣的,因此在服用时不能压碎、折断或咀嚼。这种类型的药物需要与牛奶或食物一起服用,以尽量减少胃肠道不适。患有消化性溃疡病的患者禁止使用这种药物。对正在服用抗凝血药华法林的患者,必须谨慎使用这种药物。
1/ 有机硝酸酯:有助于舒张冠状动脉,从而增加心肌的血流量。
2/ 钙通道阻滞剂:阻止 Ca+ 进入心肌细胞,并使心脏的血管开放。
3/ β 受体阻滞剂:- 阻断 β1 受体,从而抑制心脏的活动。- 导致血压略微下降,从而通过降低心脏的工作量来保护心脏。
1/ I 类:钠通道阻滞剂 2/ II 类:β-肾上腺素能阻滞剂 3/ III 类:钾通道阻滞剂 4/ IV 类:钙通道阻滞剂 5/ 其他抗心律失常药
这种类型的药物能够杀死或阻止致病微生物在体内增殖。细菌杀灭剂在遇到抑菌剂时将停止对生长微生物的作用。
注意:需要完成整个治疗过程。
1/ 阿米巴杀灭剂
- 氯喹:阿的平
- 依氟鸟氨酸:奥尼狄
- 碘喹啉:优碘新
- 氟拉唑酮:呋喃唑酮
- 羟基氯喹:羟基氯喹
- 甲氟喹:疟疾灵
- 甲硝唑:灭滴灵
- 戊胺:喷他咪丁,戊胺
2/ 氨基糖苷类
该组药物作用于致病微生物的蛋白质合成途径,从而杀死细菌,主要用于治疗顽固的革兰氏阴性菌(铜绿假单胞菌、大肠杆菌、变形杆菌、克雷伯菌和肠杆菌)。这种药物的剂量和频率肾功能必须在分配这种类型的药物时进行监测,因为其剂量和频率强烈依赖于肾功能。
警告: : 这些药物会损害肾脏和耳朵。
- 阿米卡星:Amikin
- 妥布霉素:庆大霉素
- 卡那霉素:Kantrex
- 新霉素:各种
- 奈替米星:Netromycin
- 链霉素
- 妥布霉素
3/ 抗真菌药 - 全身性和抗真菌药 - 局部性 全身性抗真菌药干扰真菌的细胞壁合成途径或蛋白质合成途径。肝功能必须严格控制,因为任何这些药物都会损害肝脏。
- 全身性抗真菌药
- 两性霉素 B:AmBisome
- 氟康唑:氟康唑
- 氟胞嘧啶:安可本
- 灰黄霉素:灰黄霉素,灰黄霉素
- 伊曲康唑:斯皮诺诺
- 酮康唑:尼挫尔
- 甲硝唑:灭滴灵
- 制霉菌素:制霉菌素,尼司他丁
- 特比萘芬:兰美抒
- 抗真菌药 - 局部性
- 杆菌肽:各种
- 环孢素:洛普罗,潘拉
- 克霉唑:洛米特林,迈克西乐
- 依康唑:Spectazole
- 龙胆紫
- 酮康唑:尼挫尔
- 咪康唑:米康唑,得舒汀,Zeasorb-AF
- 制霉菌素:制霉菌素,尼司他丁
- 氧康唑:氧康唑
- 替康唑:泰乐唑
- 硫康唑:Vagistat
- 萘替芬:达克宁
- 特比萘芬:兰美抒
- 甲硝唑:各种
- 4/ 头孢菌素
这种药物通常用于破坏细菌,因为它影响致病微生物中细胞壁的合成。需要谨慎地为对青霉素敏感的患者或肾功能受损的患者开处方。
- 头孢克洛:头孢克洛
- 头孢氨苄:Duricef,Ultracef
- 头孢唑啉:安塞夫,Kefzol
- 头孢西丁:Mefozin
- 头孢地尼:奥美尼
- 头孢托拉:Spectracef
- 头孢吡肟:Maxipime
- 头孢他啶:Fortaz
- 头孢替坦:Cefizox
- 头孢克肟:Suprax
- 头孢哌酮:Cefobid
- 头孢噻肟:Claforan
- 头孢替坦:Cefotan
- 头孢泊肟:Vantin
- 头孢丙烯:Cefzil
- 头孢曲松:罗氏芬
- 头孢呋辛:Ceftin
- 头孢拉定:Feflex
- 头孢拉啶:Anspor,Velosef
- 5/ 红霉素
红霉素的功能,无论是杀死细菌还是阻止细菌生长,都取决于使用的剂量。它可以通过与细菌核糖体的 50S 亚基结合来阻止致病微生物的蛋白质合成。这种药物会损害胃,并用于对青霉素过敏的患者。
- 阿奇霉素:希舒美
- 克拉霉素:Biazin
- 红霉素:碱基,Staticin,Emgel,Illotycin,酯化盐,乙基琥珀酸盐,硬脂酸盐,乳酸盐
- 红霉素+磺胺异噁唑:Pediazole
- 替利霉素:凯特克
- 6/ 青霉素衍生物
青霉素衍生物能够通过在复制过程中阻止致病微生物的细菌细胞壁合成来杀死细菌。副作用:: 皮疹、荨麻疹、过敏性休克。储存:必须充分摇匀并冷藏。
- 阿莫西林+克拉维酸:阿莫西林克拉维酸
- 阿莫西林:各种,阿莫西林
- 氨苄西林:各种
- 巴氨苄西林:Geocillin
- 氯西林:Tegopen
- 双氯西林:Dynapen,Dycil
- 美洛西林:美洛西林
- 萘夫西林:Unipen,Nafcil
- 奥硝唑:Bactocil,Prostaphlin
- 青霉素 G:苄星青霉素
- 青霉素 B,普鲁卡因:Wycillin
- 青霉素 VK:各种
- 哌拉西林:哌拉西林
- 替卡西林:替卡西林
- 7/ 四环素
它是一种广谱抗生素。它与 30S 核糖体亚基结合以阻止蛋白质合成,从而阻止致病微生物的生长。谨慎:会导致永久性牙齿变色、光敏性、过度晒伤。怀孕后半期禁止服用,也不要给 8 岁以下的儿童服用。过期的四环素会损害肾脏。
- 多西环素:Vibramycin
- 米诺环素:Minocin
- 四环素:各种
- 8/ 抗病毒药
- 9/ 磺胺类
- 10/ 抗疟药
- 11/ 抗结核药
- 12/ 抗胆碱能药物
1/利尿剂
2/β阻滞剂
3/ACE抑制剂
4/钙通道阻滞剂
5/其他药物
(甲状腺素)。这类药物参与体内多种重要过程,包括蛋白质合成、脂类和碳水化合物代谢、能量储存、体温等。
- 左甲状腺素:优甲乐,优速克
- 利奥甲状腺素:赛治
- 甲状腺片剂:甲状腺素
这类药物通常用于治疗哮喘。它通过支气管或控制呼吸的中枢神经系统影响呼吸系统。有助于舒张支气管平滑肌。
常见药物
- 沙丁胺醇:喘息定,万托林
- 氨茶碱
- 肾上腺素:肾上腺素
- 异丙托溴铵:阿托伐他汀,异丙肾上腺素
- 利瓦布特罗:舒利迭
- 麻黄碱:喘定
- 吡布特罗:美息敏
- 沙美特罗:希舒乐
- 特布他林:喘定
- 茶碱:多种
这类药物迫使心脏以更快的速度和更强的收缩跳动,从而导致血管收缩,增加循环系统中的血液量并升高血压。
| 多巴胺 | 多巴胺 | 仅供注射,严禁与碳酸氢钠混合 |
| 多巴胺 | 多巴胺 | 仅供注射,严禁与其他药物混合 |
泻药用于帮助便秘患者排便。
1/刺激性泻药:用于引起肌肉活动增加
2/盐类泻药:用于在小肠产生渗透压效应。
3/容积性泻药:用于增加粪便的体积和湿润粪便的内容物。
4/粪便软化剂:降低肠液内容物的表面张力
5/润滑剂:用于防止水分从肠道中吸收出来。
- 1/抗焦虑药
苯二氮卓类药物是众所周知的治疗焦虑的药物,但效果轻微。它用于减少焦虑,但不能阻止惊恐发作。抑制大脑边缘系统和皮质下水平的中枢神经系统。选择性5-羟色胺再摄取抑制剂(SSIRs)如百忧解和帕罗西汀也被广泛使用。治疗用途:减轻焦虑,镇静,催眠。
警告:可能引起嗜睡,不能与酒精类物质一起使用。
- 2/抗抑郁药
这类药物通过改变大脑中化学递质的浓度来发挥作用。效果通常需要 2 周或更长时间才能显现。抗抑郁药物的基本类型
A/三环类抗抑郁药:丙咪嗪(多虑平)非常有名,有片剂、胶囊和注射剂类型。它可以缓解抑郁,但不能完全治愈。它主要通过阻止大量去甲肾上腺素在体内传递来影响去甲肾上腺素系统,并恢复适当的平衡。然而,多虑平也会影响其他神经递质系统,如 5-羟色胺。这种药物通常需要 2 到 8 周才能显现其效果。
副作用:患者可能会出现视力模糊、口干、便秘、排尿困难、嗜睡、体重增加(平均至少 13 磅)、性功能障碍。谨慎:三环类药物过量服用会导致致命。
B/单胺氧化酶抑制剂:阻断单胺氧化酶抑制剂,该酶会分解去甲肾上腺素和 5-羟色胺。MAO 抑制剂似乎与三环类药物有等效的效果,但副作用更少。警告:致命副作用 - 患者禁止食用含有酪胺的食物,如奶酪、红酒、啤酒或某些非处方药,可能与 MAO 抑制剂相互作用并导致死亡。
C/选择性 5-羟色胺再摄取抑制剂 (SSRI):影响 5-羟色胺的突触前再摄取系统。氟西汀 (百忧解) 是这类药物中的一种知名药物。它的有效性与其他抗抑郁药基本相同,但在青少年中自杀率略有下降。警告:副作用:身体激动、性功能障碍、性欲低下、失眠、胃肠道不适。
- 3/抗精神病药
(神经阻滞剂,主要镇静剂):这些药物通过干扰多巴胺神经递质系统、血清素、谷氨酸系统来帮助人们更清晰地思考,并减少幻觉和妄想。治疗用途:治疗精神病和精神分裂症。
副作用:不必要的身体症状(嗜睡、视力模糊、口干)、帕金森氏症状、无动症(面部表情呆滞、运动缓慢、言语单调)、迟发性运动障碍(舌头、面部、嘴巴、下巴的无意识运动,或舌头伸出、脸颊鼓起、嘴巴噘起、咀嚼运动)。
| 维生素 | 名称 | 治疗用途 |
|---|---|---|
| A | 视黄醇 | 保持眼睛健康,帮助骨骼和牙齿生长,并预防感染 |
| B1 | 硫胺素 | 用于分解碳水化合物以获得能量 |
| B2 | 核黄素 | 分解脂肪以获得能量,组织呼吸 |
| B3 | 烟酸 | 用于分解脂肪、碳水化合物、蛋白质。参与激素和脂肪的合成过程 |
| B6 | 吡哆醇 | 用于分解多肽链以产生能量 |
| B9 | 叶酸 | 功能是制造新的细胞,如红细胞 |
| B12 | 氰钴胺 | 功能是制造新的细胞,如红细胞,对神经系统有益 |
| C | 抗坏血酸 | 帮助愈合受伤的组织,预防感染,促进铁吸收 |
| D2 | 麦角钙化醇 | 帮助身体吸收钙并维持血液中的 Ca+ 水平,以及促进骨骼形成 |
| D3 | 胆钙化醇 | 帮助身体吸收钙并维持血液中的 Ca+ 水平,以及促进骨骼形成 |
| E | 示例 | 保护维生素 A 和多不饱和脂肪免受氧化(抗氧化剂) |
| K1 | 示例 | 血液凝固 |
| K3 | 甲萘醌 | 血液凝固 |
Reifman, Noah. 药剂师技术人员认证复习. 第 9 版. 美国: AuthorHouse, 2011. 61-88. 打印版。
"模块 6:健康评估". 亚利桑那州卫生服务部. 亚利桑那州卫生服务部. 网页. 2012 年 11 月 20 日. <http://www.azdhs.gov/azwic/documents/local_agencies/trainingmanual_pdf/module_6.pdf>.
Durand, V. Mark 和 David H. Barlow. 异常心理学要点. 第 5 版. 12. 贝尔蒙特: Wadsworth Pub Co, 2009. 打印版。由于市场上药物批准数量处于历史最低水平,药物开发一直在受到多方面的压力。药物开发人员必须在开发过程开始之前就认识到这种趋势。不幸的是,对于一些公司而言,资源本来就有限,不像其他公司那样能够轻松地进行研究和开发。
随着时间的推移,监管要求和承诺不断增加,影响了试验规模和试验时间。这导致药物开发的总成本总体上升。FDA 现在要求进行更多样本试验才能获得批准,这是为了确保安全性和有效性。
由于临床试验数量的增加,在国外进行试验变得必要。不幸的是,每个国家都有自己的一套药物法规,进一步增加了试验的难度。
向监管机构进行电子提交已成为一些国家(如美国)的强制要求。向电子通用技术文档 (eCTD) 的过渡将很快成为强制性和首选的提交方式。虽然 eCTD 确实有很多好处,但发展中国家必须迅速适应该系统,这进一步减缓了药物的开发过程。
与药物作用和副作用相关的药物管理和吸收水平是该过程的重要组成部分。
许多化合物在进入体内后会产生重大影响,但只有极小一部分具有成为有用药物的潜力。在湿实验室合成或从自然界中提取的外来化合物,必须能够适应生物体细胞以有效发挥作用,而不会造成任何严重伤害。候选药物必须对其靶标具有强效的调节作用,因为我们拥有适合其靶标的特性。
为了使药物有效,它需要在以合理剂量服用时结合足够数量的靶蛋白。决定药物有效性的一个因素是药物与其靶标之间相互作用的强度。与某些靶分子结合的分子通常被称为配体。随着配体浓度的增加,配体分子逐渐占据更多的靶结合位点,直到基本上所有可用的位点都被占据。配体与其靶标结合的这种趋势由解离常数 Kd 测量。
Kd = [R][L]/[RL]
其中 [R] 是游离受体的浓度,[L] 是游离配体的浓度,[RL] 是受体-配体复合物的浓度。解离常数值是衡量候选药物与靶标之间相互作用强度的指标;该值越低,相互作用越强。当结合位点的浓度远小于解离常数时,游离配体的浓度等于结合位点的一半时的浓度即为解离常数。
然而,有时在利用候选药物对活细胞或组织进行生物测定的情况下,会使用另一种方法来确定药物的效力。在这些情况下,会测量 EC50 浓度。这是产生最大生物反应的 50% 所需的候选药物浓度。对于作为抑制剂的候选药物(例如钠通道阻滞剂),术语 IC50 用于描述抑制剂减少反应至其在不存在抑制剂的情况下值的 50% 所需的抑制剂浓度。
IC50 = Ki(1 + [S]/KM)

(Ki 被称为抑制常数;KM 是底物 S 的米氏常数。天然底物浓度越高,抑制酶至特定程度所需的药物浓度就越高。
IC50 和 EC50 值是在评估所需生物靶标的活性时,候选药物效力的重要衡量指标。通常,药物靶标是庞大的蛋白质家族中的一员,这些蛋白质的性质相似,这在开发靶向药物时极具挑战性。
除了分子作用于特定靶分子外,有效的药物还必须具有其他特性。例如,它必须易于给药并以足够的浓度到达其靶标以发挥作用,药物分子在其到达靶标的途中会遇到各种障碍。以下特性是药物在体内生命周期的四个基本阶段。
1. 吸收
2. 分布
3. 代谢
4. 排泄
药物可以以小片剂的形式口服,必须能够在肠道的酸性条件下存活,然后被吸收通过肠上皮细胞。一些最常见的给药方式包括口服(吞服阿司匹林片剂)、肌内注射(在手臂肌肉中注射流感疫苗)、皮下注射(在皮肤下注射胰岛素)、静脉注射(通过静脉接受化疗)或透皮(佩戴皮肤贴剂)。药物在吸收过程中面临着巨大的挑战,因为肝脏和人体药物给药的可变性很大。有一套规则告诉我们什么时候可能发生不良吸收。
1. 分子量 > 500 g/mol
2. 氢键供体数量大于 5
3. 氢键受体数量大于 10
4. 分配系数大于 5(衡量分子溶解细胞膜的趋势的一种方法)
肝脏的挑战在于,肝脏会过滤掉大部分药物,使其无法进入血液。代谢酶会分解药物,使其失效。这会导致大部分药物无法到达靶器官,或没有任何作用。为了绕过这种情况,引入了许多不同的吸收方法。(2)
一旦药物被吸收,下一步就是药物的分布。大多数情况下,血液是药物的主要运输方式。一旦药物进入血液,它就会分布到不同的体液和组织中。这是导致人体各个器官出现各种副作用的一步。但需要注意的是,一些器官的防御系统比其他器官更强大。例如,中枢神经系统具有强大的血脑屏障,可以保护大脑免受危险的毒药或病毒的侵害。
在药物在体内的分布过程中,另一个挑战是它除了与靶器官的受体结合外,还会与其他分子结合。药物可能会与患者血液中存在的化合物发生反应,最终导致药物失效,或产生不良副作用。此外,由于化合物会散布到全身,非靶器官具有特殊细胞和独特受体,最终可能会发生反应并与药物结合。如果药物与受体结合并激活或激活细胞的功能,可能会导致不良副作用,这些副作用可能具有潜在的危害或致命性。(2)
药物在全身分布并完成其特定生物学任务后,会被分解或代谢。代谢通常发生在肝脏中。肝脏是持续但受控活性的场所。进入血液的一切物质都会直接被输送到肠道。在肠道中,分子和物质会在化学和物理上代谢。
一旦药物被吸收并执行其特定的生物学任务,它必须被转化为一种能够通过物理方式排泄的物质。肝脏利用化学代谢物解毒药物的成分,然后通过尿液或粪便排出体外。
1. Berg J, Tymoczko J, Lubert S: 生化,第七版
2. 美国卫生与公众服务部,NIH 出版物,由设计医学,2006 年 7 月重印
1. Berg J, Tymoczko J, Lubert S: 生物化学,第7版 2. Singh, Harjit. 药物开发挑战。制药。<http://www.pharmafocusasia.com/strategy/drug_development_challenges.htm> 3. 设计药物 美国国立卫生研究院

骨质疏松症是一种骨骼疾病,会导致骨折和恶化。双膦酸盐,也被称为二膦酸盐,是治疗和降低患病风险的药物。它是化学家用来寻找骨质疏松症药物的关键途径之一。双膦酸盐能够与一种称为羟基磷灰石的天然矿物质结合,这种矿物质可以构成骨骼的 70%。阿仑膦酸盐(福善美)、依班膦酸盐(邦力维)、利塞膦酸盐(阿克铁)和唑来膦酸(瑞可坦)是三种用于治疗骨质疏松症的药物。

双膦酸盐是合成化合物,自 19 世纪中叶以来一直用作运河和灌溉系统中的水软化剂。它们在治疗骨质疏松症中的效率可归因于该化合物的生物学和物理化学特性的排列。双膦酸盐具有令人印象深刻的细胞选择性层,这些层构成作用时间长的药物,能够安全地抑制破骨细胞对骨骼的吸收。与骨骼表面上的羟基磷灰石结合后,大约 50% 的双膦酸盐会粘附在那里,骨骼结合阿仑膦酸盐的半衰期估计约为 10 年。没有粘附的药物剂量会迅速排泄,而双膦酸盐的低细胞渗透性会将其暴露在其他组织中的风险降到最低,从而降低发生副作用的可能性。这种对骨骼进行基本靶向的特性对于治疗骨质疏松症中随后的细胞活动非常有用。在骨骼吸收过程中,破骨细胞会附着在双膦酸盐上,因此将无法吸收骨骼。



流感病毒,也称为流感,是一种比普通感冒更严重的疾病,而且具有高度传染性。病毒可以通过空气、咳嗽、打喷嚏以及直接接触传播。它影响所有年龄段的人群,但幼儿比成人更容易感染这种疾病。这是一种季节性疾病,在全球蔓延,每年造成数千人死亡。在流感病毒靶点神经氨酸酶的情况下,对 N2 和 B 型神经氨酸酶的晶体学分析对于理解活性位点可塑性如何影响对目前商业药物扎那米韦(瑞乐扎®)和奥司他韦(达菲®)出现明显抗性变体至关重要。扎那米韦是市面上第一种药物(10),它显示出较差的口服生物利用度(2%),这可能是由于其在生理 pH 值(11)下的两性离子特性。需要口服吸入。在奥司他韦(12)的情况下,羧酸盐是一种乙酯前药,因此使该分子呈阳离子,从而增强口服吸收(75%)。所需的羧酸盐在宿主体内通过酶促水解产生(13)。此外,扎那米韦的胍基被简化为氨基,奥司他韦的甘油部分被重新设计为疏水醚,这导致结合后酶构象发生变化。这种修饰可能导致对奥司他韦产生耐药性的问题。
骨质疏松症:Biota 公司发现了扎那米韦,这是一种有助于治疗骨质疏松症和类似疾病的药物。它是同类药物中第一种上市的商业药物,可以为骨骼疾病患者服务,但不幸的是,由于其 2% 的低生物利用度,证明其疗效较低。由于扎那米韦的两性离子特性,人体无法完全吸收药量。

Biota 应该研究开发扎那米韦的酯类以提高其口服生物利用度,但他们出于正当理由没有这样做。在 Biota 发现这种药物期间,吉利德集团制备了奥司他韦的游离羧酸盐和胍类类似物,这是一种前药。为了模拟扎那米韦,吉利德集团用胍基取代了扎那米韦的氨基,最终导致口服生物利用度降低。因此,该公司得出的结论是,扎那米韦的前药形式可能不像其原始形式那样具有生物利用度,这可能是由于胍基的一些缓解作用。显然,在生产药物时,将一个参数与大局相一致可能会对另一个参数产生负面影响。扎那米韦的许可被授予葛兰素史克公司,用于最终开发;尽管 Biota 或 GSK 努力提高该药物的口服生物利用度,但这款药物的销量还是低于口服有效的奥司他韦。

流感:在动物和人身上的实验中,吸入扎那米韦剂量可以有效治疗流感。虽然扎那米韦是一种酯类前药,但如果口服,该药物将无法成功地在体内分布,因此,无法对抗传染病。由于这个问题,人们进行了多次尝试,通过改变该化合物的结构来改善扎那米韦的药理特性。

骨质疏松症:在发现扎那米韦之后,为了改善骨骼疾病,吉利德集团开发了一种名为奥司他韦的药物。这种新药与扎那米韦的功能相同,但疗效更高,销量是扎那米韦的 3 倍。这种药物比扎那米韦更成功,因为它的羧酸盐是一种乙酯前药,增强了该分子的阳离子性,因此增加了口服吸收(75%)。

流感:研究人员试图改善扎那米韦的口服摄入,通过去除羧酸,并将其改变为碳环骨架,重新设计了吡喃的结构化合物,从而发现了靶向流感病毒的有效抗病毒产品(奥司他韦),该产品可以口服服用。总的来说,统计数据显示,在日本,奥司他韦的用量比扎那米韦高约 90 倍。
Kozarich, John W. "疾病的生物化学:迫切寻求耦合。"Annu. Rev. Biochem. (2009): 55-63. 网页。
Colman, Peter M. "新型抗病毒药物和耐药性。"Annu. Rev. Biochem. (2009): 95-118. 网页。
图像来源:wikimedia-commons

在美国,食品药品监督管理局(FDA)要求在潜在药物可以大规模用于人体之前,必须证明其有效且安全。对于要由相对健康的人服用的一种候选药物,这一要求尤为重要。这些试验在 FDA 批准候选药物用于一般用途之前,测试了候选药物的有效性和潜在副作用。
临床试验至少分三个阶段进行。
第一阶段:在第一阶段,少量(10-100)健康志愿者服用药物,进行初步的安全研究。这些志愿者接受不同剂量的药物,并接受监测以观察中毒迹象。
第二阶段:在第二阶段,药物候选物的疗效将在少量可能从药物中获益的人群中进行测试。还将获得更多关于药物安全性的数据。此类试验通常受控并双盲进行。
第三阶段:在第三阶段,将在更大的人群(数千人)中进行类似的研究。本阶段将牢固地确立药物候选物的疗效,并检测可能最终出现在一小部分接受治疗的受试者身上的副作用。
只有在所有三个阶段完成后,药物才能被批准供公众临床使用。
临床研究旨在增加与疾病或病症的治疗、诊断和预防相关的医学知识。进行临床研究的一些常见原因包括
- 评估一种或多种治疗疾病、综合征或病症的干预措施
- 寻找预防疾病或病症初次发生/复发的方法
- 检查识别疾病或该疾病风险因素的方法
- 探索通过支持性护理改善慢性病患者的舒适度和生活质量的方法
临床试验通常由主要研究者领导,主要研究者通常是医生。临床研究有一个研究团队,可能包括医生、护士、社会工作者和其他医疗保健专业人员。临床研究可以在许多地方进行,包括医院、大学、医生办公室和社区诊所。地点取决于谁在进行研究。
受控研究涉及一组接受安慰剂的个体和另一组接受实际药物的个体。这些研究也往往是双盲的,因此受试者和研究人员都不知道哪些受试者在治疗组,哪些受试者在对照组。这种方法可以防止在试验过程中出现偏差。在试验完成后,受试者的分配将被解封,并比较两组的结果。在第二阶段试验中,通常会调查多种剂量,以确定哪些剂量似乎没有严重的副作用,哪些剂量似乎有效。
临床试验往往非常昂贵。成本可能从数千万美元到数亿美元不等。将创建大量记录和文档,并在随后进行整理以提交给 FDA。目前估计,开发一种药物的全部成本为 4 亿至 8 亿美元。
一项研究表明,从实验室到患者手中,一种药物大约需要 15 年时间。
1. Berg J, Tymoczko J, Lubert S: 生化,第七版
2. 克罗恩病和结肠炎基金会:临床试验 101
3. 临床试验(政府网站):了解临床研究
管制物质法 (CSA) 包含许多法律,规定了某些物质在美国的生产和分配方式。该法案也被称为 1970 年全面药物滥用预防和控制法案,由美国缉毒局 (DEA) 管理,DEA 在州际和国际层面调查非法生产受控物质。[1]
根据 CSA 第 812 节,受控物质分为五个附表。每种受控物质根据其滥用潜力、在美国接受的医疗治疗和安全性以及其可能造成身体和心理依赖的可能性,被归入附表 I、II、III、IV 或 V。
- 该药物或其他物质具有高度滥用潜力。
- 该药物或其他物质目前在美国的治疗中没有接受的医疗用途。
- 在医疗监督下使用该药物或其他物质缺乏接受的安全性。[2]
美国任何地方都不接受附表 I 药物的处方。
附表 I 药物的一些例子包括海洛因、大麻、摇头丸和 LSD。这些药物含有致幻物质、鸦片类药物(如乙酰美沙酮)或任何鸦片类衍生物,包括其异构体、酯类、盐类或其他化学组合物。[3]
- 该药物或其他物质具有高度滥用潜力。
- 该药物或其他物质在美国治疗中具有目前接受的医疗用途,或在严格限制下具有目前接受的医疗用途。
- 滥用该药物或其他物质可能导致严重的心理或生理依赖。[4]
附表 II 药物的处方只能由持证从业人员亲笔签发。在加利福尼亚州,附表 II 处方必须使用新的防篡改处方单,并且不允许重复填充。通常这些处方是为 30 天的供应量开具的。[5]
附表 II 麻醉剂的一些例子包括羟考酮、安非他明盐和哌甲酯。更知名的品牌药物包括奥施康定、派瑞可西、阿得拉和利他能。这些药物含有任何鸦片、阿片类药物或甲基苯丙胺的衍生物。
- 该药物或其他物质的滥用潜力低于附表 I 和 II 中的药物或其他物质。
- 该药物或其他物质在美国治疗中具有目前接受的医疗用途。
- 滥用该药物或其他物质可能导致轻度或低度身体依赖或高度心理依赖。[6]
附表 III 药物的一些例子包括维可丁、含有可待因的泰诺和舒缓贴。这些药物含有某些化合物或镇静剂、哌甲酯和麻醉药物的混合物。虽然附表 III 麻醉剂中混有一些附表 II 药物,但 DEA 允许每 100 毫升最多 1.8 毫克的可待因。其他一些规定包括每 100 毫升最多 500 毫克的鸦片,以及每 100 毫升最多 50 毫克的吗啡化合物。[7]
- 与附表 III 中的药物或其他物质相比,该药物或其他物质的滥用潜力较低。
- 该药物或其他物质在美国治疗中具有目前接受的医疗用途。
- 与附表 III 中的药物或其他物质相比,滥用该药物或其他物质可能导致有限的身体依赖或心理依赖。[8]
附表 IV 药物的一些例子包括阿普唑仑、地西泮、劳拉西泮和卡里索普罗多。更知名的品牌名称包括安定、安定、阿提汎和索玛。[9] 这些药物含有巴比妥类、美索卡品、苯巴比妥或甲丙氨酯,通常用于治疗焦虑和失眠。
- 该药物或其他物质与附表 IV 中的药物或其他物质相比,具有较低的滥用潜力。
- 该药物或其他物质在美国治疗中具有目前接受的医疗用途。
- 与附表 IV 中的药物或其他物质相比,滥用该药物或其他物质可能导致有限的身体依赖或心理依赖。 [10]
附表 V 麻醉品的例子包括罗布麻咳嗽糖浆和其他镇咳药。许多这些药物含有可待因、二氢可待因、乙基吗啡、苯乙酰吗啡或鸦片的组合。然而,混合物中允许的活性麻醉药物含量有限。例如,每 100 毫升混合物中只能含有 100 毫克鸦片。 [11]
天然产物是由活生物体产生的化学物质。它存在于自然界,并具有可以被研究以支持药物发现和药物设计的生物学效应。即使可以通过全合成制备,某种物质也被认为是天然产物。 [12]
天然产物的来源包括植物、细菌、海洋环境和动物毒液。
嗜粪动物是专门在动物粪便中繁衍的生物。这些领地物种会向附近的真菌吐出有毒化学物质。科学家和研究人员利用这些信息,专门寻找对某些真菌有毒的化学物质,这些化学物质可能对感染的人类有潜在的危险。
蓝细菌是植物状生物,生活在潮湿和潮湿的环境中。这些物种已被证明是癌症和细菌细胞杀手的来源。例如,化合物隐藻素-8 可以破坏一系列肿瘤的支架。另一种名为 majusculamide C 的分子专注于真菌,使其有可能用于治疗人类的真菌相关疾病。
滤食动物是粘附在岩石和珊瑚上的生物。这些物种相互竞争食物和其他自然资源。科学家和研究人员发现,从长远来看,其中一些强效化学物质可用于治疗癌症和其他致命疾病。
最终,这些天然产物已被科学地利用来创造想要和不想要的效果。简而言之,许多这些化学物质在医药领域展现出光明的前景。 [13]
蓝细菌,或蓝绿藻,是植物状生物,生活在潮湿和潮湿的环境中。蓝细菌是光合生物,可以利用阳光产生能量。这些微生物可以在称为藻华的菌落中找到,其历史可以追溯到地球上最古老的化石。它们构成了世界上最大的细菌群体之一,与许多人类和动物疾病有关。当细菌在几天内快速生长时,就会形成藻华。这些藻华会使清澈的水变得浑浊。蓝细菌藻华很危险,因为它们会消耗掉生长水域中存在的所有氧气,从而杀死生活在那里的其他植物和动物。一些蓝细菌甚至产生了一些已知的最强效的天然毒素,没有解药。
然而,这些物种已被证明是癌症和细菌细胞杀手的来源。例如,化合物隐藻素-8 可以破坏一系列肿瘤的支架。另一种名为 majusculamide C 的分子专注于真菌,使其有可能用于治疗人类的真菌相关疾病。
滤食动物是粘附在岩石和珊瑚上的生物。这些物种相互竞争食物和其他自然资源。滤食动物使用水生觅食方法来获取食物。科学家和研究人员发现,从长远来看,滤食动物中发现的一些强效化学物质可用于治疗癌症和其他致命疾病。
滤食动物包括海绵,它是最简单的多细胞动物。海绵产生大量的毒素,它们要么释放到水中,要么出现在其表面。这些毒素会抵御任何以海绵为食的捕食者。接触这些毒素的症状包括:接触部位发红、疼痛、刺痛、瘙痒、肿胀、肿块、恶心,甚至昏厥。
尽管有毒性,海绵在对抗传染病和癌症方面已显示出巨大希望。科学家们已能够从海绵中提取抗病毒、抗癌和抗肿瘤化合物,以创造许多药物。例如,在 1950 年代,化学家使用海绵化合物创造了一种治疗疱疹的药物,称为阿昔洛韦(伐昔洛韦),以及一种治疗非霍奇金淋巴瘤的药物,称为阿糖胞苷(胞苷阿拉伯糖苷)。 [14]
最终,所有这些天然产物已被科学地利用来创造想要和不想要的效果。简而言之,许多这些化学物质在医药领域展现出光明的前景。

海鞘属于滤食动物类,对癌症药物的医学研究有很大贡献。海鞘也被称为海鞘,看起来只是小小的彩色斑点,但对制药界来说比人们想象的要多得多。例如,在西印度群岛的珊瑚礁中,一种名为桶形海鞘的海鞘有助于对抗癌症。这种海洋动物,正如伊利诺伊大学的肯·赖因哈特所发现的那样,包含了一种名为海鞘素的天然物质,用于制造名为 Yondelis™ 的抗癌药物。尽管这种药物仍处于初步阶段,还需要进一步研究,但实验室测试已经证实它可以杀死癌细胞,适合人类服用。
最近,哈佛大学的埃利亚斯·J·科里发现了在实验室合成海鞘素的方法。这很重要,因为为了生产一克药物,需要收获超过一吨的海鞘;这显然是一个无效的过程。随着合成感兴趣的天然产物的能力,科学家们正在推动利用天然产物生产有效药物治疗疾病的进步。 [15]

HEALTH AND HUMAN SERVICES,</ref>]] 紫杉醇是一种用于治疗癌症的药物。它来源于一种名为太平洋红豆杉(Taxus brevifolia)的树的树皮和针叶,提取紫杉醇后该树会死亡。另一方面,虽然它是对抗癌症最有效的药物之一,但从大量的红豆杉树皮中提取的紫杉醇含量很少(大约 1200 公斤树皮能产生 10 克纯紫杉醇)。由于这种药物的生产对环境产生了负面影响,因此紫杉醇的制造引起了人们对红豆杉种群的生态影响的担忧。
紫杉醇通过阻止癌细胞复制来发挥作用。它通过在细胞分裂过程中与微管结合,阻止它们分解来做到这一点。由于微管通常在细胞分裂后被分解,因此微管的存在会阻止细胞分裂成子细胞。 [16]

毒蛙碱是一种存在于厄瓜多尔蛙(Epipedobates tricolor)皮肤上的分子,具有镇痛作用,这意味着它是有效的止痛药。尽管研究证明毒蛙碱比吗啡更有效的止痛药,但毒蛙碱是有毒的,少量剂量就会杀死大型生物。因此,毒蛙碱不太可能在医药市场上出售。
毒蛙碱通过结合烟碱型乙酰胆碱受体(nAChR)结合位点发挥作用。当烟碱型乙酰胆碱受体与神经递质结合时,它会释放多巴胺和去甲肾上腺素,使生物体对疼痛不敏感。 [17]

肉毒梭菌是一种细菌,会产生一种称为神经毒素的毒素,它们会导致食物中毒。在 1960 年代后期,科学家艾伦·斯科特在猴子身上测试了肉毒杆菌毒素 A 型(BTX-A),发现 BTX-A 可用于治疗斜视(眼睛没有对齐的一种状况)。
肉毒杆菌毒素最广为人知的用途是,注射到皮肤中,可以减少皱纹和眉间纹。许多名人接受肉毒杆菌毒素(Botox,是肉毒杆菌毒素的简称)注射,以使面部皮肤光滑。[18]
- ↑ “管制药物法。”管制药物法案 (CSA)。HG.org 全球法律资源,无日期。网络。<http://www.hg.org/control.html>.
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ “监管信息 - 管制药物法案。”美国食品药品监督管理局。美国卫生与公众服务部,无日期。网络。<http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ http://www.mbc.ca.gov/licensee/rx_form_requirements.html
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ “监管信息 - 管制药物法案。”美国食品药品监督管理局。美国卫生与公众服务部,无日期。网络。<http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ “DEA 药物管制 - 管制药物类别。”DEA 药物管制 - 管制药物类别。美国缉毒局,无日期。网络。<http://www.deadiversion.usdoj.gov/schedules/index.html>.
- ↑ “监管信息 - 管制药物法案。”美国食品药品监督管理局。美国卫生与公众服务部,无日期。网络。<http://www.fda.gov/regulatoryinformation/legislation/ucm148726.htm>.
- ↑ http://www.thefreedictionary.com/Natural+product
- ↑ Berg,Jeremy M.,编辑 (2002),生物化学 (第 6 版) 纽约市,纽约州:W.H. Freeman 和公司,
- ↑ http://www.allthesea.com/Sea-Sponge.html
- ↑ Davis,Alison,博士,(2006),设计药物:来自大自然的药物,过去和现在:海洋药物 (NIH 出版物编号 06-474):美国卫生与公众服务部,
- ↑ http://www.research.vt.edu/resmag/1999resmag/taxol.html
- ↑ http://www.chm.bris.ac.uk/webprojects2002/jjones/Content/Epibatidine.htm
- ↑ http://www.animalresearch.info/en/medical/diseasesresearch/Botulinum
解释药物如何与细胞膜相互作用以及与药物相互作用相关的膜结构特征。
许多药物需要穿过一个或多个细胞膜才能到达其作用部位。所有细胞膜的共同特征是磷脂双分子层,大约 10 纳米厚。跨越该双分子层或附着在外层或内层小叶上的是糖蛋白,它们可能充当离子通道、受体、中间信使 (G 蛋白) 或酶。细胞从细胞外液中获取分子和离子,从而形成持续的进出流动。细胞膜有趣的地方在于,相对浓度和磷脂双分子层会阻止必需离子进入细胞。因此,为了使药物穿过细胞膜,必须解决这些问题。通常,这是通过协助扩散或主动运输来完成的。在协助扩散中,相对浓度用于进出运输。主动运输使用能量 (ATP) 将分子和离子进出细胞。[1]
细胞信号通过称为信号转导的过程穿过细胞膜。当特定信息遇到细胞外表面并直接与受体接触时,就会发生这三个步骤的过程。受体是一种特殊分子,它接收来自环境的信息,并将信息传递到细胞的各个部分。接下来,一个连接开关分子,转导器,将信息传递到细胞内部。最后,信号被放大,因此导致细胞执行特定功能。这些功能可能包括移动、产生更多蛋白质,甚至发出更多信号。[2]
药物穿过细胞膜最常见的方法是被动扩散。药物分子将在其浓度梯度下降的情况下扩散,而无需细胞消耗能量。但是,膜具有选择性渗透性,因此它对不同药物分子的扩散速率具有不同的影响。通过膜中的转运蛋白进行协助扩散,也可以提高扩散速率。有两种类型的转运蛋白执行协助扩散,通道蛋白和载体蛋白。[3]
主动运输是一个需要能量的过程。药物分子逆浓度梯度运输,大多数使用的蛋白质是载体蛋白,而不是通道蛋白。主动运输也有两种类型。
初级主动运输直接利用能量将分子跨膜运输。有时载体蛋白可以是产电泵。[4]
次级主动运输或协同运输也使用能量将分子跨膜运输。然而,它与初级运输的不同之处在于,它没有直接与三磷酸腺苷 (ATP) 耦合;相反,它利用泵出细胞离子的电化学势差。[5]

MDR 代表多药耐药,指的是大多数生物体内细胞表面膜中的一种大型整合蛋白泵。它们的主要功能是监控通过细胞膜的化学物质,并将可能危害细胞健康的化学物质排出。通常它们对细胞的生长有益,但当生长不受欢迎时,例如细菌感染或癌细胞扩散,MDR 泵就会成为问题。MDR 泵提供了适应和防御某些抗生素的细胞能力。细胞利用 MDR 泵来抵抗环境,研究人员不仅需要合成影响靶细胞的抗生素,还需要能够穿透细胞的防御。 MDR 泵通过利用称为 ATP 的能量包来排出不需要的毒素而被激活。
MDR 泵存在于人体各个部位,包括大脑、消化道、肝脏和肾脏。这些泵在人体中执行特定的功能,例如将激素等分子转运进出细胞。
研究人员认为,植物随着时间的推移进化出了阻止 MDR 泵的化学物质,本质上与抗生素需要达到的效果相同。该理论通过敲除特定细菌细胞(金黄色葡萄球菌)的 MDR 泵基因进行了验证。然后将细菌暴露于从黄连根中提取的抗生素黄连碱。黄连碱已被证明在金黄色葡萄球菌存在的情况下通常无效。然而,它对没有 MDR 泵的基因改造细菌非常有效。当细菌也暴露于被认为能抑制细菌 MDR 泵的黄连碱提取物中时,这些细菌在黄连碱存在的情况下也被杀死。
MDR 泵是多药耐药泵,是微生物细胞表面膜中的一种大型蛋白质。它们用于监控进入的化学物质,并排出可能危害细菌的化学物质。为了使泵工作,必须打开闸门。当泵打开时,它可以吸收药物。泵的细胞质闸门关闭,使药物可以被泵出细胞。
MDR 泵可以赋予微生物以抵抗抗生素的形式的致命力量,这些微生物是细菌。虽然细菌 MDR 基因与那些基因并不完全相同,但所有泵的三维形状都是相同的,它们执行类似的任务:防止有害分子进入细胞内部。许多 MDR 泵赋予了对首次用于治疗致病性酵母和寄生虫的药物的抗性。对用于治疗酵母菌感染的抗真菌药物的耐药性正在迅速增加,影响着免疫系统受损的艾滋病患者。 MDR 是一种称为蛋白酶抑制剂的抗艾滋病药物沙奎那韦。MDR 泵也可能转运“正常”物质。在哺乳动物中,MDR 泵被放置在肠道中,以便面对面地遇到潜在的有害物质。
研究人员正专注于通过使用 MDR 泵来对抗癌症。干细胞,即尚未分化的细胞,在其膜中只有很少的 MDR 泵。因此,这些干细胞对杀死癌细胞的药物非常敏感。化疗的一个主要缺点是骨髓毒性,这会使人体无法抵抗致命感染。科学家和研究人员设计了一种用 MDR 泵连接无防御干细胞的方法。这样做,干细胞可能有机会抵抗杀死人体肿瘤所需的化疗。 [1]
肯塔基大学列克星敦的药理学家玛丽·沃尔最近的发现表明,MDR 泵在妇女怀孕期间无法正常工作。基本上,患有妊娠期肝内胆汁淤积症(ICP)的孕妇的 MDR 泵畸形,这会危及发育中的胎儿。对此的推测原因是怀孕期间雌激素和其他妊娠激素的影响。
文件:Mdr pump.jpg 科学家和研究人员认为,药物对服用药物的人中的不到一半产生效果。这部分是由于生活方式和环境因素,但主要是由于基因变异。制造细胞色素 P450 蛋白的基因负责药物在体内的作用。这些蛋白质代谢人体产生的激素和外来激素。由于每个人都拥有不同的基因组序列,基因编码的蛋白质也存在差异。这种基因变异会影响细胞色素 P450 的反应方式以及药物如何产生效果。最终,细胞色素 P450 蛋白处理我们服用的许多药物,因此解释了个人对药物的不同反应。 [2]
http://upload.wikimedia.org/wikipedia/commons/4/42/DoseResponse.png
上图是剂量反应曲线,它告诉我们药物(x 轴)在多大程度上引起身体的反应(y 轴)。蓝色曲线和绿色曲线之间的区域是预期效果,绿色曲线右侧的区域是副作用。科学家研究药物及其对人体的反应之间的关系。就像图像显示的那样,当没有投入药物时,一开始没有效果。然后研究人员在系统中添加更多药物以观察对身体的影响。如果服用过多药物,身体将达到过量服用点,并可能面临副作用。这张图片展示了研究人员在处理药物时寻找的合适剂量。 [3]
Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) New York City, NY: W.H. Freeman and Company
Davis, Alison Davis. Medicines By Design. The Office of Communications and Public Liaison. 2006.
流感病毒是呼吸道感染的主要罪魁祸首,通常被称为“流感”。流感病毒的结构包括一个被衣壳包围的核蛋白(RNA)中心、一个脂质包膜,以及在其表面上的两种关键蛋白的尖峰:血凝素和神经氨酸酶。大约 80% 的尖峰由血凝素组成。血凝素的功能是使病毒与宿主细胞结合。它起着将病毒粘附到宿主细胞上的作用,以引起感染。另一种蛋白质称为神经氨酸酶,覆盖了表面的其余部分。较少的神经氨酸酶并不影响其重要作用。神经氨酸酶有助于促进新形成的病毒分子从宿主细胞中释放。从这个角度来看,血凝素是将病毒与宿主细胞结合的锚点,神经氨酸酶是诱发病毒感染的触发器。由于神经氨酸酶对流感病毒非常重要,科学家们已经开发出了抑制该蛋白质并防止流感病毒感染宿主细胞的方法。
基于结构的设计一直是药物设计的主要方法之一。利用 X 射线晶体学和核磁共振(NMR)两种关键技术,它们帮助确定了分子的结构。有关特定目标的三维结构等信息有助于指导药物的创建,因为基于结构的设计允许观察目标与药物之间的相互作用。
最近,研究表明,药物与其靶标的天然配体的相似性与抗性增加的障碍之间存在正相关性。换句话说,药物的设计需要考虑与靶标的天然配体在物理和化学上的相似性,这将使所述疾病和靶标更难产生耐药性。
发现用于治疗流感病毒的神经氨酸酶抑制剂是基于结构的药物设计早期实例之一。流感药物的目标是基于病毒的神经氨酸酶。该酶的作用是释放病毒的后代,以便在体内传播并感染其他细胞。神经氨酸酶抑制剂是根据病毒神经氨酸酶的晶体结构数据创建的。神经氨酸酶抑制剂的作用是化学破坏病毒的受体,从而阻止病毒复制。神经氨酸酶抑制剂必须在症状出现后的 48 小时内服用。它们不会“杀死”流感病毒,而只是减缓病毒复制速度,直到免疫系统能够更容易地将其摧毁。因此,它们可以减轻流感病情的严重程度和持续时间。

流感病毒在体内的传播通过以下步骤减缓:(1)病毒在存在神经氨酸酶抑制剂的情况下进入细胞。(2)一旦进入细胞,神经氨酸酶抑制剂就会附着在病毒上。(3)流感病毒仍然可以使用宿主来复制自身。(4)然而,抑制剂阻止病毒离开细胞,从而阻止感染其他细胞。(5)病毒变得无效并在细胞内死亡。
目前,已经发现十种神经氨酸酶,并确定了它们的结构:流感 A 的 N1-N9 和流感 B 的 B 型神经氨酸酶。已批准供公众使用的四种药物是扎那米韦、奥司他韦、金刚烷胺和金刚乙胺。
扎那米韦、奥司他韦、金刚烷胺和金刚乙胺在抑制的流感病毒类型、给药途径和年龄组的批准使用方面有所不同。然而,扎那米韦和奥司他韦的副作用和成本与金刚烷胺和金刚乙胺不同。据报道,服用扎那米韦和奥司他韦的患者出现神经紧张、焦虑、注意力困难和头晕等副作用的频率较低。此外,扎那米韦和奥司他韦比利巴韦更贵,而利巴韦比金刚烷胺更贵。虽然金刚烷胺和金刚乙胺都是被广泛用于治疗流感 A 的药物,但扎那米韦和奥司他韦是新的替代方案。与扎那米韦和奥司他韦不同,金刚烷胺和金刚乙胺是化学相关的抗病毒药物,它们可以对抗流感 A 病毒,但不能对抗流感 B 病毒。然而,这些流感抗病毒药物不能替代疫苗,而是补充剂。
实验发现,这两种药物根据它们的摄入方式和与靶标的结合亲和力具有不同的抗性。奥司他韦以片剂形式口服,而扎那米韦以吸入剂形式服用。奥司他韦是一种前药,这意味着它在进入体内之前是无活性的。两种药物都发现,根据上述方法,它们的效果更好。对于抑制剂,氨基或胍基取代了神经氨酸酶的 C4-羟基。改变取代基可以在化学和物理上优化结合效力。据科尔曼博士说,“对神经氨酸酶……扎那米韦和奥司他韦羧酸盐的抑制效力的损失,越不类似于底物,则越低”。[4]
更多研究表明,有充分的证据支持这样一种假设,即类似于靶标的天然底物和配体的药物在抑制药物耐药性方面更加成功。事实上,病毒需要能够与其自身的底物结合,并能够区分自身底物与药物的底物。如果药物类似于病毒自身的底物,那么病毒将难以区分,因此,抵抗力会降低。此外,这就是为什么要开具多种药物和多种剂量的原因。使用“药物混合物”、剂量和给药途径将带来更好的效果。
流感病毒细胞表面上的两种重要蛋白质是凝集素血凝素蛋白和酶唾液酸酶。凝集素血凝素具有 3 个浅唾液酸结合位点,而酶唾液酸酶在一个口袋中具有一个活性位点。低分子量抑制剂的深活性位点使唾液酸酶成为比血凝素更具吸引力的抗流感药物靶标。
流感病毒与唾液酸残基结合。因此,病毒通过粘附在碳水化合物的表面来进入宿主细胞。与糖结合的病毒蛋白称为血凝素。一旦病毒穿透细胞膜,一种名为神经氨酸酶的病毒蛋白就会裂解唾液酸残基的糖苷键,使病毒能够感染宿主细胞。神经氨酸酶抑制剂是唾液酸的类似物,它们会阻断神经氨酸酶的活性位点,并在宿主细胞和流感病毒包膜的表面留下未裂解的唾液酸残基。病毒血凝素与未裂解的唾液酸残基结合,减少了病毒向其他细胞的传播。
- ↑ http://sciencenotes.ucsc.edu/9701/pdf/mdrartII.pdf
- ↑ Berg,Jeremy M.,编。 (2002),生物化学(第 6 版)纽约市,纽约州:W.H. Freeman and Company
- ↑ 戴维斯,艾莉森·戴维斯。按设计生产药物。通信和公众联络办公室。2006 年。
- ↑ 科尔曼,彼得·M. “新型抗病毒药物和耐药性”,'生物化学年度综述',2009 年。
- 科尔曼,彼得·M. “新型抗病毒药物和耐药性”,'生物化学年度综述',2009 年。
阿普唑仑(也称为赞安诺)是一种用于治疗焦虑症和惊恐症的精神活性药物。它属于一类名为苯二氮卓类药物的药物,其作用是减少大脑中异常的兴奋。阿普唑仑可以有多种形状和大小。它们可以是片剂、缓释片、口崩片,也可以是液体形式。阿普唑仑用于治疗焦虑症和惊恐症(突然且意外的极度恐惧和不安的攻击,以及对这些想法的不安)。阿普唑仑属于一类名为苯二氮卓类药物的药物,其作用是减少大脑中异常的兴奋。
阿普唑仑有时也用于治疗抑郁症、旷野恐惧症(广场恐惧症)和经前综合征。这种药物可能被用于其他用途;请咨询您的医生或药剂师了解更多信息。
阿普唑仑现在是辉瑞公司的一部分,但最初由 Upjohn 公司发布。它在美国专利 3,987,052 的保护下,该专利于 1969 年 10 月申请,并于 1993 年 9 月到期。阿普唑仑于 1981 年发布。它发布时,第一个获批的适应症是惊恐症。
一位名叫大卫·希恩的年轻精神科医生建议使用名为 DSM-III 的新区分。因此,DSM-III 在焦虑症的分类中创建了广泛性焦虑症 (GAD) 和惊恐症之间的区别,以便将阿普唑仑推向市场。换句话说,这是希恩为宣传阿普唑仑的使用而采取的一种方式。从他的研究中,希恩知道惊恐症在人群中很普遍,而且对苯二氮卓类药物有反应。因此,他有了主意,并建议 Upjohn 公司将阿普唑仑用于惊恐症的营销,这既可以涵盖新的诊断领域,又可以强调这种药物的独特效力。他基本上是在寻找独特的方式来展示阿普唑仑有多棒。
随着他的临床研究的继续,希恩描述了第一批他用阿普唑仑测试的患者是如何说这种药物非常有效。他们基本上都对阿普唑仑产生的作用印象深刻。几个月后,当美国食品药品监督管理局批准阿普唑仑时,阿普唑仑很快就被抢购一空,他们获得了巨额利润。然而,与许多其他药物类似,许多发表在医学文献中的报告指出,服用阿普唑仑会导致严重的戒断症状,包括精神病、癫痫发作和严重的反弹焦虑。
阿普唑仑在缓解中度至重度焦虑和惊恐发作方面有效。由于对耐药性、依赖性和滥用的担忧,它不再是这些发作的一线防御药物。医生限制患者使用阿普唑仑的时间为 4 到 10 周,并且只允许将其分配给没有药物耐药性或依赖性史的人。
在怀孕期间服用阿普唑仑存在潜在风险。它会导致胎儿药物依赖和产后戒断症状。它还会导致新生儿无力和呼吸问题。已知阿普唑仑会从人乳中排出并传给婴儿。阿普唑仑会导致婴儿体重减轻和嗜睡。
阿普唑仑是一种来自苯二氮卓类药物的中央神经系统药物,其作用机制尚不清楚。预计它通过与中枢神经系统中不同的立体异构体受体结合而发挥作用。阿普唑仑也已被证明具有抗抑郁特性,这通常不是常规苯二氮卓类药物的常见特性。
阿普唑仑会引起多种副作用。最常见的副作用是
- 嗜睡
- 头晕
- 头痛
- 疲劳
- 眩晕
- 易怒
- 健谈
- 注意力不集中
- 口干
- 唾液分泌增加
- 性欲或性能力改变
- 恶心
- 便秘
- 食欲改变
- 体重变化
- 排尿困难
- 关节疼痛
除了这些常见的副作用外,它有时还会产生更多副作用,例如
- 呼吸短促
- 癫痫发作
- 看到或听到不存在的东西(幻觉)
- 严重的皮疹
- 皮肤或眼睛发黄
- 抑郁
- 记忆问题
- 困惑
- 言语障碍
- 行为或情绪异常变化
- 考虑伤害或杀死自己或试图这样做
- 协调或平衡问题
阿普唑仑通过 CYP3A4 代谢。氟西汀、酮康唑、西咪替丁、奈法唑酮、利托那韦、普罗泊芬、红霉素和伊曲康唑都是 CYP3A4 抑制剂组合的例子。这些抑制剂基本上会延迟阿普唑仑的肝脏清除,从而导致阿普唑仑过度积累。
据报道,伊米普拉明和去甲替林同时服用阿普唑仑片剂,剂量高达每天 4 毫克,平均分别增加了 31% 和 20%。众所周知,联合口服避孕药会降低阿普唑仑的清除率。这会导致阿普唑仑的血浆浓度增加和蓄积。
此外,卡瓦根和阿普唑仑如果结合使用,会对彼此产生很大影响,因为会导致半昏迷状态。金丝桃属是金丝桃科约 400 种开花植物的属,可以降低阿普唑仑的血浆浓度,并降低其治疗效果。然而,从大局来看,酒精是最重要的和最常见的相互作用。这是由于联合用药的想法。酒精和苯二氮卓类药物(如阿普唑仑)联合使用对彼此具有协同作用。这种反应会导致严重的镇静、行为改变和中毒。因此,通常认为,服用酒精和阿普唑仑的量越多,相互作用就越严重。
通用名:(阿普唑仑)
- 口服片剂:0.25 毫克、0.5 毫克、1 毫克、2 毫克
- 口服片剂,崩解片:0.25 毫克、0.5 毫克、1 毫克、2 毫克
- 口服片剂,缓释片:0.5 毫克、1 毫克、2 毫克、3 毫克
阿普唑仑 Intensol(口服溶液):1 毫克/毫升
Niravam:口服片剂,崩解片:0.25 毫克、0.5 毫克、1 毫克、2 毫克
Xanax:口服片剂:0.25 毫克、0.5 毫克、1 毫克、2 毫克
Xanax XR:口服片剂,缓释片:0.5 毫克、1 毫克、2 毫克、3 毫克
http://www.nlm.nih.gov/medlineplus/druginfo/meds/a684001.html http://www.medicinenet.com/alprazolam-oral/page2.htm http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000807 http://en.wikipedia.org/wiki/Alprazolam 氨甲喋呤

氨甲喋呤是一种有效的嵌入式抗肿瘤药物。它在治疗成人急性白血病和恶性淋巴瘤方面有效,但在治疗实体瘤方面无效。它在化学疗法中常与其他抗肿瘤药物联合使用。
注射剂(静脉注射)
- 黑色大便
- 咳嗽或声音嘶哑
- 发烧或寒战
- 排尿疼痛或困难
- 皮肤上出现针尖大小的红点
- 嘴唇、舌头或口腔内出现疮、溃疡或白点
- 腹部疼痛或压痛
- 视力模糊
- 心跳或脉搏快、强或不规则
- 心悸
- 癫痫发作
- 眼睛或皮肤发黄


DNA缠绕在称为组蛋白的基本蛋白质周围,并且被紧密地盘绕或超螺旋。在 DNA 序列被 DNA 加工酶加工之前,必须将其解开或松弛。拓扑异构酶 II 或 TopoII 是一种可以加工 DNA 的酶。TopoII 会暂时断裂 DNA 链,并允许两端在酶内自由旋转。这允许 DNA 解开。在 DNA 被加工后,酶会重新连接这两个端点。
氨甲喋呤通过抑制 Topo II 酶杀死癌细胞。它稳定了酶与断裂的 DNA 链复合物的时间足够长,以便酶-DNA 复合物完全分解。当 DNA 被破坏时,癌细胞最终会被杀死。
"氨甲喋呤。" Scott Hamilton CARES Initiative。无名氏。网页。2012 年 12 月 7 日。<http://chemocare.com/chemotherapy/drug-info/Amsacrine.aspx> Grove,WR,CL Fortner 和 PH Wiernik。"临床药学。"临床药学。7 月-8 月。(1982):320-26。印刷版。

阿司匹林属于非甾体抗炎药(NSAID)家族。阿司匹林已被使用了几千年。据信它最早在公元前400年由希腊医生希波克拉底使用,他用柳树的树皮和叶子为病人缓解疼痛和发烧。柳树富含一种叫做水杨苷的物质,这种物质在公元1832年由德国化学家查尔斯·格尔哈特转化为水杨酸。最后在公元1897年,德国化学家菲利克斯·霍夫曼发明了阿司匹林,导致拜耳在公元1899年发布阿司匹林。科学家们使用X射线晶体学来确定COX(环氧合酶)酶的3D结构,这帮助他们研究了酶的形状以及像阿司匹林这样的药物如何阻断它。
COX抑制剂
[edit | edit source]许多药物属于NSAID(非甾体抗炎药)家族,如Advil、布洛芬和Aleve。这些药物的功能类似,它们都阻断一类称为环氧合酶(缩写为COX)的酶。这些药物阻断两种相似的酶,称为COX-1和COX-2。COX-2在感染或损伤时触发免疫反应和肿胀。阻断COX-2是可取的,因为它可以减少肿胀、发烧、疼痛等。这些药物(如阿司匹林)的问题是它们也会阻断COX-1。COX-1产生保护胃壁的分子,称为前列腺素。因此,阻断COX-1会导致溃疡,因为胃酸能够消化没有保护的胃壁。这对长期使用阿司匹林治疗关节炎等疾病来说是一个重大问题。长期使用阿司匹林的这些副作用导致美国每年约有16,500人死亡。[1]
仅COX-2抑制剂的开发
[edit | edit source]

多年来,科学家一直在思考像阿司匹林这样的COX抑制剂是如何起作用的,因此他们使用了X射线晶体学和其他生物物理技术来确定环氧合酶的三维结构。酶的结构对于理解其功能至关重要。可视化单个酶的结构折叠和弯曲使科学家能够考虑可能与酶结合的潜在抑制剂。随着COX-1和COX-2结构的解析,人们发现了一种新的药物,它可以阻断COX-2,但不能阻断COX-1,称为塞来昔布。对COX-1和COX-2的解析结构表明了为什么只有COX-2被抑制。在COX-1中塞来昔布的结合位点存在异亮氨酸,而COX-2中存在缬氨酸。缬氨酸的结构具有一个类似口袋的末端,很容易与之结合,而异亮氨酸具有一个延伸的侧链结构,这使得药物无法与之结合。这使得能够制造新的药物来结合该特定靶点,而不会造成COX-1酶引起的副作用。[1]
COX-3
[edit | edit source]科学家还开发了另一种称为COX-3抑制剂的抑制剂。这种抑制剂似乎更类似于泰诺。泰诺不属于NSAID家族,因为它不是一种抗炎药。COX-3药物的未来可能会导致对泰诺的更深入了解。
合成
[edit | edit source]一种常见的阿司匹林合成方法被归类为酯化反应,如下所示[2]
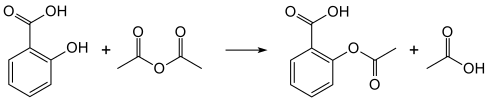
- 反应机理
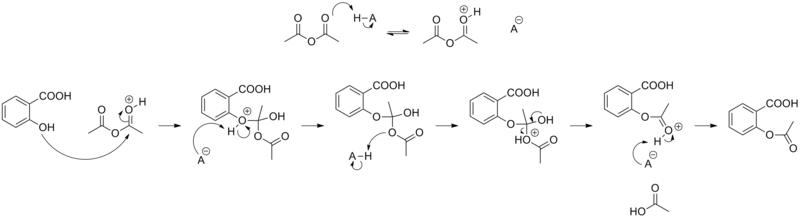


作用机制
[edit | edit source]
阿司匹林是一种抗血小板药物,可以抑制环氧合酶或COX。COX-2作为免疫反应触发炎症。通过阻断COX-2,阿司匹林起着强效的止痛剂和抗炎剂的作用。然而,COX-1酶也是血栓烷A2(TXA2)生成的關鍵酶。血栓烷TXA2会导致血小板在血管撕裂处聚集在一起,以促进血管损伤或疾病时修复。血小板的聚集会导致血凝块,从而止血并帮助修复受损的血管。阿司匹林抑制COX-1,从而抑制TXA2,从而降低血小板聚集的能力。这就是为什么阿司匹林被称为血液稀释剂或抗血小板剂的原因。
COX-1和COX-2都是具有相似结构但功能不同的环氧合酶。它们都从脂肪酸花生四烯酸生成前列腺素。然而,COX-1和COX-2会产生不同的前列腺素。来自COX-2的前列腺素与疼痛、炎症和发烧有关,而来自COX-1的前列腺素支持血小板聚集、胃肠道粘膜完整性和肾脏功能。

在阻断COX的过程中,阿司匹林上的乙酰基被水解,然后以酯的形式与丝氨酸的醇基结合。这具有阻断酶中通道的效果,花生四烯酸无法进入酶的活性位点并被转化为前列腺素。
COX-1和COX-2的氨基酸结构之间存在60%的同源性。阿司匹林通过与COX-2的Ser 516结合的方式与Ser 530结合来阻断COX-1。然而,COX-2的活性位点略大于COX-1的活性位点,因此制药公司已经能够开发出选择性的COX-2抑制剂,例如塞来昔布、罗非昔布和美洛昔康。这些药物可以抑制COX-2的生化作用,从而减少炎症,但对COX-1没有影响,可以防止胃粘膜损伤。
多态性
[edit | edit source]多态性是指一种物质能够形成不止一种晶体结构的能力。这个过程在药物成分的开发中至关重要。很长一段时间以来,只发现了一种晶体结构。然后从20世纪60年代开始,人们怀疑存在第二种晶体形式。这种第二种多晶型物首次在2005年由Vishweshwar及其同事发现。在用热乙腈进行阿司匹林和左乙拉西坦的共结晶后,基本上发现了一种新的晶体。然而,这种第二种晶体形式只在100°K时稳定,在环境温度下会恢复为第一种晶体形式。在第一种晶体形式中,存在两个水杨酸分子通过乙酰基与(酸性的)甲基质子形成中心对称二聚体,以羰基氢键的方式。此外,在最新宣称的第二种晶体形式中,每个水杨酸分子与两个相邻的分子形成相同的氢键,而不是只与一个形成。最后,两种多晶型物都形成了相同的二聚体结构,即相对于羧酸基团形成的氢键而言。
服用阿司匹林之前
[edit | edit source]发烧的儿童或青少年不应该服用阿司匹林。患有水痘或流感症状的儿童也不应该服用阿司匹林,因为阿司匹林有可能导致一种称为雷氏综合征的严重疾病,这种疾病主要发生在儿童身上。此外,如果患有以下任何疾病,成年人也不应该服用阿司匹林
- 近期有肠道、胃或颅内出血史
- 出血性疾病
- 对非甾体抗炎药过敏,如Advil、Motrin、Aleve或Orudis
如果一个人服用阿司匹林来预防中风或心脏病,那么务必避免同时服用布洛芬,因为这会增加严重出血的风险。这种药物也会伤害未出生婴儿的心脏,并可能降低出生体重或产生其他严重影响。因此,在服用阿司匹林之前,务必告诉医生是否怀孕或计划在服用该药物期间怀孕。哺乳期的母亲也必须知道,服用阿司匹林会通过母乳进入婴儿体内,并严重危害哺乳婴儿。因此,所有哺乳期的母亲在服用这种药物之前应该提前告诉医生。
药物用途
[edit | edit source]阿司匹林属于非甾体抗炎药 (NSAID) 类,自古希腊时代以来就已被使用。这种药物属于水杨酸盐类,在治疗与疼痛和炎症相关的各种疾病方面有多种用途。它通常通过减少导致炎症和疼痛的物质来起作用。通常,阿司匹林在缓解疼痛方面被认为不如布洛芬。然而,阿司匹林通常对由肌肉痉挛、腹胀或皮肤刺激引起的疼痛无效。因此,治疗轻度至中度疼痛和炎症是阿司匹林的常见用途,但阿司匹林也被用于治疗和预防心脏病、中风、胸痛和其他心血管疾病。然而,阿司匹林似乎对那些心脏病或中风风险较低的人几乎没有作用。这种理念被称为一级预防。通常在服用阿司匹林时,应该避免服用其他 NSAID,因为它们实际上会抑制药物的作用。阿司匹林有多种形式(肠溶片、咀嚼片),通常由医生开具,用于预防心血管问题。
此外,研究表明阿司匹林对癌症有很大影响,特别是它对一种被称为结直肠癌(CRC)的癌症的影响。即使是多项荟萃分析也表明,经常服用阿司匹林肯定会降低患结直肠癌的长期风险。100 多年来,阿司匹林的其他用途可能包括治疗与普通感冒相关的发烧和疼痛。基本上,阿司匹林可以作为治疗酸痛、不适、头痛和喉咙痛的来源。此外,服用高剂量阿司匹林三天内,现有的发烧和关节疼痛通常会很快治愈。患有心包炎、冠心病和急性心肌梗死的患者也可以用阿司匹林治疗。
与使用阿司匹林相关的副作用和风险因素有几个。对阿司匹林过敏、有肠道或胃出血史、出血性疾病(如血友病)以及对其他 NSAID 过敏的人应避免使用阿司匹林。但是,开处方者仍然可以在进行检查并根据患者的需要调整剂量后推荐阿司匹林。例如,患有胃溃疡、肝病、血液凝固障碍或高血压等特定医疗问题的患者可能需要进行额外的检查并调整剂量,才能安全地开具阿司匹林。此外,服用阿司匹林的患者应避免饮酒,因为饮酒会导致胃出血增加。阿司匹林的典型副作用包括胃部不适、胃灼热、嗜睡和头痛。由于阿司匹林会刺激胃和肠道的内壁,因此减少这种现象的一种有效方法是用一杯牛奶服用阿司匹林。如果出现荨麻疹或呼吸困难等副作用,应停止使用阿司匹林。
最后,在开具阿司匹林之前,应向医生告知某些药物,例如华法林。页面文本。[3]
如果您出现以下一种或多种症状,请立即就医
- 荨麻疹
- 呼吸困难
- 嘴唇、脸、舌头或喉咙肿胀
如果您出现以下一种或多种症状,请停止使用阿司匹林并致电医生
- 黑色、血性或柏油样便
- 咳血或呕吐物呈咖啡渣样
- 严重恶心、呕吐或胃痛
- 发烧持续超过 3 天
- 肿胀持续超过 10 天
- 听力问题(耳鸣)
最轻微的症状可能包括
- 胃部不适
- 胃灼热
- 头痛
- 嗜睡
Flavio,Guzmán。“抗血小板药:作用机制和概述。”药理学角落。N.p.,2009 年 24 日。网络。2012 年 12 月 7 日。
Vane,J.R 和 R.M Botting。“血栓形成研究”。血栓形成研究。110。(2003):255-58。打印。
- ↑ a b 生命的结构,美国国立卫生研究院。“生命的结构”。2007 年 7 月:46-48。
- ↑ Palleros,Daniel R. (2000)。实验有机化学。纽约:约翰威利父子公司。p. 494。 ISBN 0-471-28250-2.
- ↑ [1],附加文本。
缬沙坦/代文
描述
缬沙坦,在美国、英国和澳大利亚[1]被称为代文,在化学上被描述为 N-(1-氧戊基)-N-[[2'-(1H-四唑-5-基) [1,1'-联苯]-4-基]甲基]-L-缬氨酸。它的经验式是 C24H29N5O3,分子量为每摩尔 435.5 克[2]。
物理上,缬沙坦是一种细白粉末,可溶于乙醇和甲醇,微溶于水。代文以片剂形式供口服,含 40 毫克、80 毫克、160 毫克或 320 毫克 缬沙坦。片剂的非活性成分是胶体二氧化硅、交聚维酮、羟丙基甲基纤维素、氧化铁(黄、黑和/或红)、硬脂酸镁、微晶纤维素、聚乙二醇 8000 和二氧化钛[2]。
缬沙坦的结构式为
-
 图 1.) 缬沙坦(代文)化学结构
图 1.) 缬沙坦(代文)化学结构

机制
缬沙坦是一种血管紧张素 II 受体拮抗剂,或更常见的是,一种 I 型(AT1)血管紧张素受体[1]的血管紧张素受体阻滞剂 (ARB)。阻断血管紧张素 II AT1 受体的激活通过放松血管壁内的平滑肌细胞来扩大血管[3, 4]。该机制还减少了血管加压素和醛固酮的产生和排泄,血管加压素和醛固酮是两种导致肾脏水潴留并随后导致血压升高的激素[5, 6]。
药代动力学
缬沙坦在给药后 2-4 小时达到峰值浓度,半衰期约为 6 小时。但是,如果与食物一起服用,缬沙坦的暴露量会降低 40%,峰值浓度会降低约 50%[2]。
代谢和消除
在回收药物时,大部分药物是未改变的,但约 20% 是代谢物。发现 9% 的回收代谢物是戊酰基 4-羟基缬沙坦。从体外代谢研究中发现,CYP 2C9 同工酶是形成戊酰基 4-羟基缬沙坦的酶。口服后,约 83% 的剂量从粪便中排出,约 13% 的剂量从尿液中排出[2]。
在血压中的作用(高血压)
随着心脏跳动,血液通过血管泵出,在血管内产生一种力,这就是血压。当血压升高时,心脏必须更努力地将血液泵入血管,这会导致血管损伤,并可能导致中风、心脏病、心力衰竭、肾衰竭和视力问题[7]。
高血压的一个原因是血管紧张素 II,它会使血管收缩,从而减少血流量并增加血管内的血压[8]。因此,通过阻断血管紧张素 II AT1 受体的激活,缬沙坦通过放松和扩大血管来降低高血压,从而降低血流阻力[8]。缬沙坦还用于治疗充血性心力衰竭 (CHF) 或心肌梗死 (MI) 后[1]。
代文副作用[7]
代文对高血压患者的常见副作用:头痛、头晕、流感症状、疲劳、腹痛。
代文对心力衰竭患者的常见副作用:头晕、低血压、腹泻、关节和背部疼痛、疲劳、高血钾
代文对心脏病患者的常见副作用:低血压、咳嗽、(高血肌酐)、肾功能下降、皮疹。
影响代文的药物[9]
环孢素(健択,新妥,山德士)利尿剂(利尿片)利福平(利福定,利福特,利福美)利托那韦(诺维,卡莱特)非甾体抗炎药 (NSAID),例如阿司匹林、布洛芬(Advil,Motrin)、萘普生(萘普生,萘普生,特瑞美)、塞来昔布(塞来昔布)、双氯芬酸(Arthrotec,Cambia,Cataflam,伏特伦,Flector 贴片,潘萨德,Solareze)、吲哚美辛(吲哚美辛)、美洛昔康(Mobic)等。
[1] http://en.wikipedia.org/wiki/Valsartan#Myocardial_infarction_controversy
[2] http://www.pharma.us.novartis.com/product/pi/pdf/diovan.pdf
[3] http://en.wikipedia.org/wiki/Angiotensin_receptor_blocker
[4] http://en.wikipedia.org/wiki/Vasodilation
[5] http://en.wikipedia.org/wiki/Vasopressin
[6] http://en.wikipedia.org/wiki/Aldosterone
[7] http://www.pharma.us.novartis.com/product/pi/pdf/diovan_ppi.pdf
[8] http://www.diovan.com/info/about-diovan/what-is-diovan-and-how-it-works.jsp
[9] http://www.drugs.com/diovan.html 狄安波隆(也称为甲基雄烷酮,阿维波隆,达那波隆,甲基睾酮或甲基睾酮)是一种合成代谢类固醇,一些运动员使用它来增加肌肉的大小和力量。这类似于雄性激素睾酮。一些研究表明这种药物的效果微乎其微,但一些副作用会阻止人们使用它。
狄安波隆是在德国研发的,并于 1960 年代在美国由奇巴特殊化学品公司推出。它在美国是一种受管制物质,尽管在健美者中很流行。
美国奥运会举重队队医齐格勒博士在改变睾酮使其对人体更安全的研究中发挥了重要作用。然而,由于运动员滥用该药物,齐格勒博士成为反对运动兴奋剂的代言人。
- 肝癌风险增加
- 心脏病风险增加
- 油性皮肤、痤疮以及身体和面部毛发生长
- 肝功能障碍
- 雄激素副作用
- 对于女性,声音变粗和月经周期不规则。
概述
泰诺,也称为对乙酰氨基酚,是一种常见的止痛药。该药物以缓解头痛和退热而闻名。泰诺不被认为是 NSAID,即非甾体抗炎药。它在抑制炎症方面作用不大。
注意事项
如果你的关节因剧烈运动而疼痛,要小心。与阿司匹林或艾利相比,泰诺可能不是一个好的选择。这是因为关节疼痛是由于炎症引起的。
泰诺的主要成分对乙酰氨基酚实际上是在阿司匹林成为消费产品之前就在欧洲被发现。然而,直到麦克尼尔实验室的罗伯特·麦克尼尔才开始对其潜在的药用价值进行深入研究。在一次美国制药商协会会议上,麦克尼尔与分析镇痛药和镇静药研究所副总裁雷蒙德·康克林进行了一次非正式交谈,这一化合物 N-乙酰基对氨基酚引起了麦克尼尔的注意,并开始了他的研究工作。与阿司匹林相比,对乙酰氨基酚取得巨大成功的部分原因在于,它不会像阿司匹林那样引起胃痛。美国食品药品监督管理局(FDA)于 1955 年 6 月批准了该药物,并以 Elixir Tylenol 的名称推出。
吸收
口服对乙酰氨基酚在胃肠道中迅速且几乎完全被吸收,主要是在小肠中。这种吸收过程通过被动转运发生。相对生物利用度在 85% 到 98% 之间。
分布
对乙酰氨基酚似乎分布在大多数体液中,除了脂肪。对乙酰氨基酚的分布容积为 0.95 L/kg。一小部分对乙酰氨基酚与血浆蛋白结合,并且结合量仅在与过量相关的血浆浓度中略有增加。即使在相对较高的浓度下,硫酸盐和葡萄糖醛酸盐代谢物也不与血浆蛋白结合。
代谢
对乙酰氨基酚主要在肝脏中代谢,涉及三个主要独立的途径:1. 与葡萄糖醛酸结合;2. 与硫酸盐结合;3. 通过细胞色素氧化。
排泄
对乙酰氨基酚主要通过形成葡萄糖醛酸和硫酸盐结合物从体内消除。
泰诺的作用机制 泰诺,即对乙酰氨基酚,属于一类称为非阿片类镇痛药的止痛药。非阿片类镇痛药通过抑制一种称为环氧合酶(COX)的酶发挥作用。COX 是一种催化剂,可以将细胞壁中的一种脂肪酸,即花生四烯酸,转化为一种称为前列腺素的物质。前列腺素会引起疼痛、炎症和发烧。它在周围神经系统中,在损伤部位引起细胞损伤后的疼痛和炎症。它们通过影响下丘脑来升高体温。通过阻断 COX 和随后在中枢和周围神经系统中产生的前列腺素,泰诺可以减轻发烧和炎症。泰诺与其他非阿片类药物的不同之处在于,它在周围神经系统中对 COX 的阻断作用并不大。它似乎主要通过一种或多种机制在中枢神经系统中减轻疼痛,可能部分是通过抑制一种称为 COX-3 的 COX 形式。因此,它被认为是一种弱镇痛药,不具有抗炎特性。
泰诺和阿司匹林结构的相似性 这两种分子之间的第一个相似之处是它们的苯骨架。这两种分子在苯环上都具有与反应性氧和反应性氢连接的基团,这有助于它们的分子酸性。阿司匹林具有羧酸,而泰诺含有酚。这两种分子的酸性有助于类似的生理活性,即缓解疼痛。

- 泰诺 - 上图

- 阿司匹林 - 下图
有些人服用泰诺后可能会出现过敏反应。如果出现以下一种或多种症状,建议去看医生。
- 荨麻疹
- 呼吸困难
- 面部、嘴唇、舌头或喉咙肿胀
- 恶心
- 胃部不适
- 瘙痒
- 食欲不振
- 尿液颜色变深
- 陶土色粪便
- 黄疸(皮肤或眼睛发黄)
如果与某些其他药物一起服用,泰诺可能会发生剧烈反应。可能与泰诺发生反应并产生严重副作用的药物包括:
- 抗生素
- 避孕药/激素替代疗法
- 血压药
- 癌症药物
- 降胆固醇药物
- 关节炎药物(包括金注射剂)
- 艾滋病毒/艾滋病药物
- 精神疾病药物
- 癫痫药物
对乙酰氨基酚诱发肝坏死 尽管对乙酰氨基酚在治疗剂量下是安全的,但较高剂量会导致肝坏死。对乙酰氨基酚中毒占美国和英国所有急性肝功能衰竭病例的近一半。这源于以下步骤:1. 对乙酰氨基酚被细胞色素 450 代谢激活为一种反应性物质,这种物质会消耗谷胱甘肽,谷胱甘肽是一种抗氧化剂,可以防止活性氧物质(如自由基和过氧化物)对重要细胞成分造成的损害。2. 谷胱甘肽的损失以及肝细胞中活性氧和氮物质的增加会发生坏死性变化;3. 由于氧化应激增加,线粒体通透性发生变化。4. 线粒体通透性因额外的氧化应激而发生变化,导致线粒体膜电位的丧失,以及线粒体合成 ATP 能力的丧失。5. ATP 的丧失将使肝细胞无法再正常运作,导致坏死。某些细胞因子和趋化因子等炎症介质可以改变毒性。一些已被证明可以改变氧化应激。此外,现有数据支持细胞因子、趋化因子和生长因子参与肝脏再生过程的启动。因此,请仔细注意对乙酰氨基酚的服用量,切勿超过最大日剂量(4000 毫克或 24 小时内服用 8 片泰诺加强型片)。如果需要更多的止痛效果,请咨询医生,考虑加入其他止痛策略或更换其他类型的止痛药。对乙酰氨基酚也存在于各种药物中,从非处方咳嗽和感冒药到处方级止痛药。请确认所有药物的成分,确保不会超过最大剂量,因为过量会导致肝坏死。如果患有肝病或大量饮酒,请完全避免服用对乙酰氨基酚!如果定期服用该药物,请注意肝损伤的迹象,包括尿液颜色变深、粪便颜色变浅、右上腹疼痛以及眼白发黄。
泰诺的其他危险 苯海拉明是泰诺 PM 中的一种成分,是一种抗组胺药,也具有镇静作用,有助于患者入睡。然而,如果人们因过度使用泰诺 PM 或在服用泰诺 PM 中含量的基础上额外服用苯海拉明而服用过量的苯海拉明,就会发生过量。这种过量会导致意识混乱、口干、疲劳、肌肉无力、头晕和胸闷。大剂量苯海拉明会导致幻觉和癫痫发作。
COX-2 和对乙酰氨基酚
从结构的角度来看,很难理解对乙酰氨基酚如何抑制 COX 的 COX 位点,因为它缺乏与精氨酸 120 相互作用的羧酸部分。早期的研究提供了一种机制方法,可以解释对乙酰氨基酚为何具有 NSAID 部分治疗活性,但缺乏抗炎和抗凝血活性。对乙酰氨基酚对其位于大脑中的一种 COX 亚型起作用。发现 COX 的两种形式证实了 COX 亚型的概念,但对 COX-2 的进一步研究发现它并没有受到对乙酰氨基酚的明显抑制。
http://www.drugs.com/tylenol.html [1] [2] [3] [4] [5] [6] [7]
"Robert L. McNeil, Jr." 化学遗产基金会主页。N.p.,n.d. Web. 2012年11月21日。<http://www.chemheritage.org/discover/online-resources/chemistry-in-history/themes/pharmaceuticals/relieving-symptoms/mcneil.aspx>.
百忧解,也称为氟西汀,是一种称为选择性血清素再摄取抑制剂(SSRI)的药物,用于治疗抑郁症、强迫症、进食障碍和惊恐发作。这种药物通过维持大脑中血清素的水平发挥作用,血清素是一种控制情绪和精神状态的神经递质。在其他情况下,百忧解用于治疗酒精中毒、注意力缺陷障碍、边缘型人格障碍、睡眠障碍、头痛、精神疾病、创伤后应激障碍、妥瑞氏症、肥胖症、性功能障碍和各种非理性恐惧。


百忧解的一些常见副作用包括抑郁、焦虑、头痛、恶心、腹泻、失眠和食欲不振。由于氟西汀对神经递质的影响,它有时会阻断其他神经递质,例如乙酰胆碱。当乙酰胆碱被阻断时,就会出现恶心。另一个危险的副作用是,选择性血清素再摄取抑制剂也会抑制一氧化氮的合成,从而导致血管收缩。这很危险,因为它会导致高血压。此外,尽管百忧解是一种抗抑郁药物,但它会导致抑郁,因为大脑中的神经化学失衡。
百忧解口服给药。服用胶囊后,胶囊在消化系统中缓慢释放。
百忧解绝不能超过处方剂量服用。神经递质和神经通路的大量失衡会导致许多副作用。孕妇不应服用百忧解,因为会导致高血压,从而导致新生儿的心脏缺陷和肺部问题。此外,百忧解不应与其他抗抑郁药一起服用,因为会导致严重的危险化学反应。最后,24岁以下的人可能会有自杀念头。
百忧解的作用机制是阻断轴突上的信号,该信号告诉轴突何时产生足够的血清素。因此,过量的血清素被传递到神经元。氟西汀在肝脏中通过CYP2D6代谢。由于代谢缓慢,它在体内的半衰期较长。由于这种缓慢的积累,过量的血清素需要一段时间才能显示出显着的作用。百忧解作为5HT2C受体的激动剂,已知与防御和攻击行为有关。人们发现,大脑中低血清素水平传统上与抑郁症相关。
神经递质是信号分子,用于在大脑中的神经元之间传递信息。神经递质在突触中的途径可以被特定的化学物质(称为神经调节剂)延长或缩短。一些神经调节剂可以帮助神经递质释放到突触中;另一些则抑制神经元对神经递质的再吸收,使神经递质保留在突触中。在某些精神疾病(如双相情感障碍、强迫症、焦虑症和抑郁症)中,神经递质在突触中的分泌和清除调节功能不正常。特别是在抑郁症和类似疾病中,神经递质血清素是受影响最严重的一种。为了治疗抑郁症,最常见的药物类型称为选择性血清素再摄取抑制剂(SSRI)。百忧解是该类药物中最广泛使用的成员之一。

百忧解通过抑制一种名为SERT的神经调节剂来调节突触中的血清素平衡,SERT将血清素泵回神经元。因此,百忧解有助于阻止血清素的再吸收,并增加突触中血清素的含量。人们认为,突触中适当的血清素含量有助于改善情绪和治疗抑郁症。
礼来公司在 1970 年代研究抗组胺药苯海拉明时发现了氟西汀。她想合成这种化合物许多衍生物,并在老鼠身上进行测试。最终,她制造出一种只抑制血清素再摄取的衍生物,她称之为百忧解。许多有争议的报道针对百忧解和礼来公司。暴力爆发、自杀和谋杀都被认为是这种药物引起的。然而,据说百忧解不会让人上瘾或习惯性。百忧解的专利在 2001 年到期后,许多通用血清素抑制剂药物开始销售。
- Johnson,George。“吸烟和药物成瘾。”背景资料。N.p. Web。2012年12月7日。
- Reichstetter,Sandra。“百忧解如何起作用。”大脑博主。N.p.,2010年19日。Web。2012年12月7日。
- http://panicdisorder.about.com/od/treatments/a/prozac.htm
- http://www.drugs.com/prozac.html
- http://www.encognitive.com/node/18210
- http://www.rxlist.com/prozac-drug.htm
- http://www.brainphysics.com/howprozacworks.php
- http://www.drugs.com/monograph/fluoxetine-hydrochloride.html
炭疽病的词源来自anthrax,这是一个希腊词,意思是煤炭。它源于希腊语中煤炭的意思,因为患有皮肤炭疽病的受害者身上会出现黑色的皮肤病变。在 1875 年,一位名叫罗伯特·科赫的德国医生首次发现了导致炭疽病的细菌。他的研究首次证明了微生物如何引起疾病。科赫揭示了炭疽病的生命周期和传播原因。他的实验还帮助人们了解了微生物在让人生病中的作用,当时人们仍在争论自发产生论和细胞学说。
此外,在 1881 年 5 月,路易·巴斯德进行了一项公开实验,以展示他关于疫苗接种的想法。为了证明这一点,他准备了两个由 25 只绵羊、几头牛和一只山羊组成的群体。不同组的动物被注射了炭疽疫苗或未接种疫苗。最终发现,未接种疫苗组的所有动物都死亡,而接种疫苗组的所有动物都存活。然后,在 1954 年,人类炭疽疫苗问世,1970 年,改良的无细胞疫苗也问世。

炭疽病是由炭疽杆菌分泌的一种二元毒素,炭疽杆菌分泌构成炭疽毒素的三个蛋白质。炭疽毒素由三个蛋白质组成,这些蛋白质单独无毒,但共同构成一个有毒的复合体。这三种蛋白质是致死因子(LF)、水肿因子(EF)和保护性抗原(PA)。致死因子和水肿因子是酶,保护性抗原是一种多功能蛋白质。
天然保护性抗原(Native PA)是一种长而扁平的蛋白质,由四个折叠结构域组成。第一个结构域(结构域 1)是一个β-夹层结构,包含四个α螺旋和两个钙离子。第二个结构域(结构域 2)由一个β-桶核心组成,并形成一个允许致死因子和水肿因子进入胞质溶胶的孔。第三个结构域(结构域 3)具有类似铁氧还蛋白的折叠结构。第四个结构域(结构域 4)由一个β-夹层结构组成,具有类似免疫球蛋白的折叠结构,用于与细胞受体结合,第四个结构域与蛋白质的其他部分接触很少。
-
 七聚体前孔结构。
七聚体前孔结构。

七聚体保护性抗原前孔是一个环,内部为空,表面是扁平的疏水结构,它是致死因子和水肿因子的结合位点。它呈蘑菇状。
-
 致死因子结构。
致死因子结构。

炭疽毒素有一个口袋,可以与宿主细胞结合,导致细胞充满液体。炭疽毒素单独无毒,但结合后会改变形状。炭疽毒素酶的活性位点是一个很有前景的药物靶点,因为这个口袋很容易被阻断。致死因子是一种依赖 Zn2+ 的内肽酶。
ANTXR1 和 ANTXR2 是两种与 PA 结合的细胞受体。ANTXR1 是一种中等长度的受体,而 ANTXR2 是一种长受体。这两种受体在细胞外结构域的各个部位都存在。

炭疽毒素在保护性抗原 (PA) 与细胞受体 ANTXR1 或 ANTXR2 中的任何一个结合并被蛋白水解激活后开始发挥作用。然后,保护性抗原形成七聚体前孔(PA 寡聚化形成中空环)。这种新的前孔与毒素的两种酶成分之一或两者结合:致死因子 (LF) 或水肿因子 (EF)。这些酶结合后形成的复合体被转移到内体中;前孔转变为跨膜孔,致死因子和水肿因子穿过孔进入细胞质。
一旦进入细胞,致死因子就会切断并使参与激活对细胞至关重要的蛋白质的蛋白激酶 (MAPKK 1 和 MAPKK 2) 失活。水肿因子抑制免疫反应并导致细胞死亡。
虽然炭疽病是一种通常影响动物的传染病,但接触到感染炭疽病的动物的人也会被感染。当人类患有这种传染病时,感染通常涉及皮肤、胃肠道或肺部。
在最常见的炭疽病感染类型皮肤炭疽病中,通常是当炭疽病孢子接触到人体皮肤上的割伤或擦伤时发生的。
当孢子进入肺部时,就会发生吸入性炭疽病。吸入炭疽病孢子并不意味着一个人会感染炭疽病。这表示他们已经接触过炭疽病。为了使吸入性炭疽病完全发展,细菌孢子必须萌发,以便实际的疾病发生。这通常需要 1 到 6 天。如果孢子萌发,它会释放有毒物质,这些物质会导致内部出血、肿胀和组织坏死。
当有人食用被炭疽病感染的肉类时,就会发生胃肠道炭疽病。
皮肤炭疽病症状在接触后 1 到 7 天开始显现
- 出现类似于昆虫叮咬的瘙痒性疮。疮可能会起泡并变黑。
- 无痛疮,但会肿胀。
- 形成痂,大约两周后干燥脱落。
吸入性炭疽病症状
- 发烧、不适、头痛、咳嗽、呼吸急促 (SOB) 和胸痛
胃肠道炭疽病症状在一周内发生,可能包括
- 腹痛
- 正常或血便
- 发烧
- 口腔溃疡
- 恶心和呕吐
炭疽病感染通常用抗生素治疗。
患有吸入性炭疽病的人会接受多种抗生素的联合治疗。医生通常首先给病人静脉注射环丙沙星和另一种药物。吸入性炭疽病的治疗通常需要约 60 天,因为孢子可能需要多达 60 天的时间才能萌发。
皮肤炭疽病用口服抗生素治疗,如多西环素和环丙沙星,持续约 7 到 10 天。
Croston, Glenn. "Anthrax Toxin Mechanism of Action ." Biocarta. N.p., n.d. Web. 30 Nov. 2011. <www.biocarta.com/pathfiles/h_anthraxPathway.asp>.
Karin, Michael, and Jin Mo Park, PhD. "Molecular Mechanism Underlying Anthrax Infection Described." UC San Diego Health System | San Diego Hospital, Healthcare . UCSD, 29 Sept. 2002. Web. 30 Nov. 2011. <http://health.ucsd.edu/news/2002/08_29_Karin.html>.
Young, John A. T.,Collier, R. John . "Anthrax toxin: Receptor binding, internalization, pore formation, and translocation." Annual Review of Biochemistry. Web. 30 Nov. 2011. <http://www.annualreviews.org/doi/pdf/10.1146/annurev.biochem.75.103004.142728>.
图片来自 Wikimedia Commons。
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002301/
http://en.wikipedia.org/wiki/Anthrax
苯乙肼是一种非选择性不可逆的 MAOI。MAOIs 代表单胺氧化酶抑制剂。它们是通过阻止单胺氧化酶分解神经递质而起作用的抗抑郁药物。这些神经递质包括血清素、去甲肾上腺素和多巴胺。由于这些神经递质没有被分解,它们会留在脑中,这有助于提高情绪。人们还认为该药物可以保护脑内的神经细胞免受谷氨酸的敏感性。这可以防止谷氨酸刺激大脑中负责抑郁和焦虑的区域。单胺氧化酶有两种类型酶。有 MAO-A 和 MAO-B。这些酶的作用是去除神经递质中的分子片段,使这些神经递质失活并可以被代谢。该药物的一些副作用包括震颤、心律不齐和癫痫发作。苯乙肼与酶结合的作用会永久性地使酶失活,并且不可逆。此外,该药物在抑制酶时不会区分 MAO-A 和 MAO-B。
苯乙肼是一种用于治疗抑郁症的药物。如上所述,这种药物属于 MAOIs(单胺氧化酶抑制剂)。使用这种药物时,会增加人体维持心理平衡所需的一些天然物质的含量。
这种药物不会恢复人们的抑郁症状况,它只是帮助控制症状。服用这种药物的平均时间可能是 4 周或更长。服用这种药物时,如果你的状况好转,你不能停药。你必须咨询你的医生,以便他/她可以减少你的常规剂量。如果你停药,你会经历呕吐、恶心、噩梦和虚弱等症状。
与许多其他药物一样,苯乙肼在服用这种药片时会有一些副作用,应该引起注意
- 虚弱
- 口干
- 头痛
- 便秘
- 身体部位震颤
- 胸痛
- 持续性心悸
- 恶心
- 夜间出汗
- 呕吐
- 体重增加
- 疼痛
http://publications.nigms.nih.gov/medbydesign/medbydesign.pdf
http://www.mayoclinic.com/health/maois/MH00072
http://online.factsandcomparisons.com
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000573/
AHFS® 消费者药物信息。© 版权所有,2011 年。美国医院药剂师协会,Inc。
佩普奇是一种由默克公司生产的药物,其通用名为法莫替丁(Famotidine)。它属于组胺 H2 受体拮抗剂,化学名为 2-[4-[2-(氨基磺酰亚氨基甲基)乙基硫代甲基]-1,3-噻唑-2-基]胍。法莫替丁用于治疗和预防胃和肠溃疡。法莫替丁还用于治疗胃酸分泌过多以及胃酸逆流至食道引起烧心的疾病,例如胃食管反流病 (GERD)。该药物有悬浮液、注射液和片剂形式。15% 到 20% 的血浆中佩普奇与蛋白质结合。口服的佩普奇片剂和悬浮液吸收不完全,其生物利用度为 40-45%。佩普奇通过肾脏 (65-70%) 和代谢 (30-35%) 途径排泄,唯一确定的代谢产物是 S-氧化物。其半衰期为 2.5-3.5 小时。该药物通过抑制胃酸分泌起作用。这是通过竞争性抑制组胺 H2 受体实现的。这些受体位于壁细胞中,负责胃中 HCl 的分泌。当肥大细胞被激发时,它们会释放 H2 底物,然后与受体位点结合并产生 HCl。该药物与 H2 竞争受体位点,以防止结合,从而防止胃酸释放。该药物的一些严重副作用包括癫痫发作、心律不齐和腹泻。

历史
[edit | edit source]山之内制药株式会社开发了佩普奇(法莫替丁)。在 80 年代中期,默克公司获得了佩普奇的许可,并由默克和强生之间的合资企业进行销售。使用 2-胍基噻唑环取代咪唑环。最终,许多研究表明法莫替丁的活性是西咪替丁的 30 倍。
在 1999 年,佩普奇 RPD 口溶片上市。它们无需吞咽。然后在 2001 年,仿制药上市。例如,Fluxid(施维雅)或 Quamatel(盖德昂·里希特有限公司)。
此外,在美国还有一种名为佩普奇全效的产品。然而,在英国,这种产品被称为佩普奇双效。佩普奇全效是一种将法莫替丁与抗酸剂组合在咀嚼片中的产品,以改善其相对较慢的起效时间。
然而,佩普奇也有一些副作用。例如,法莫替丁在胃中低 pH 值下溶解性差,生物利用度低 (50%)。法莫替丁与抗酸剂联合使用可促进这些药物向壁细胞壁受体的局部递送。因此,许多研究人员正在寻找开发片剂的创新制剂,例如胃滞留药物递送系统。这是因为这种类型的药物已知在胃中停留更长时间,从而提高药物的生物利用度。
医疗用途
[edit | edit source]在世界不同国家,某些法莫替丁制剂可以非处方 (OTC) 获取。例如,在美国,10 毫克和 20 毫克的片剂制剂,有时甚至与传统的抗酸剂联合使用,都可以作为非处方药获取。然而,如果个人决定服用更大剂量的药物,他们应咨询医生以获得处方。在这种情况下,处方将使他们能够从非处方药中获得所需的足量法莫替丁。
佩普奇通常给予接受手术的患者。他们通常在手术前服用佩普奇,以预防术后恶心并降低发生吸入性肺炎的风险。此外,佩普奇也给予服用非甾体抗炎药 (NSAID) 的一些患者。服用佩普奇基本上可以预防他们患上消化性溃疡,因为这种药物通常是质子泵抑制剂的替代品。一个有趣的事实是,佩普奇不仅给予人类,也给予动物,例如狗。它们被给予患有胃酸反流的狗。
许多服用法莫替丁的人也使用这种药物与 H1 受体拮抗剂联合治疗和预防荨麻疹。荨麻疹是由急性过敏反应引起的。已发现法莫替丁通过阻断组胺可以减轻慢性心力衰竭的虚弱效应。
总而言之,佩普奇或法莫替丁可以作为一种有用的药物,尤其是在正确使用的情况下。
佩普奇的副作用
[edit | edit source]- 胃肠道
胃肠道副作用包括腹泻 (1.7%) 和便秘 (1.2%)。呕吐、恶心、腹痛、厌食和口干很少见。也报道过临床意义不明确的高胃泌素血症。
- 神经系统
大发作性癫痫发作;精神障碍,在有后续追踪的病例中可以改变,包括幻觉、意识混乱、激动、抑郁、焦虑、性欲下降;感觉异常;失眠;嗜睡。在肾功能减退的患者中,很少报道过抽搐。
- 肝脏
肝脏副作用包括轻度肝功能指标升高。这些升高的临床意义尚不清楚。黄疸和胆汁淤积性黄疸很少见。也报道过药物性肝炎病例。
- 心血管
心血管副作用包括心搏量和心脏产量的降低,在以前有心脏功能障碍的患者中可能具有临床意义。也报道过各种心律失常,包括心动过缓、心动过速、房室传导阻滞(包括房室阻滞)和心悸。在肾功能受损的患者中,极少数报道过 QT 间期延长,这些患者的法莫替丁剂量/给药间隔可能未得到适当调整。
- 过敏反应
过敏反应副作用包括过敏反应、血管性水肿、眼眶或面部水肿、荨麻疹、皮疹、结膜感染、毒性表皮坏死松解症(非常罕见)、多形性红斑和史蒂文斯-约翰逊综合征。
- 肾脏 肾脏副作用包括罕见间质性肾炎。
- 内分泌
内分泌副作用包括可逆性高泌乳素血症和乳房发育的抗雄激素作用。
- 皮肤
皮肤副作用包括脱发、痤疮、瘙痒、皮肤干燥和潮红。报道过至少一例接触佩普奇的工人的接触性皮炎。
- 血液
已报道过血液副作用,包括中性粒细胞减少症。罕见情况下,也报道过可逆性血小板减少症、粒细胞缺乏症、全血细胞减少症和白细胞减少症。
- 呼吸系统
报道过呼吸系统副作用,包括支气管痉挛和间质性肺炎。
- 骨骼肌肉
骨骼肌肉副作用包括肌肉疼痛,包括肌肉痉挛和关节痛。
- 其他
其他副作用包括耳鸣、发热、无力、疲劳和味觉障碍。报道过至少一例与使用佩普奇相关的体温过高。
参考文献
[edit | edit source]http://online.factsandcomparisons.com
http://www.mamashealth.com/stomach.asp
http://en.wikipedia.org/wiki/Famotidine
http://www.drugs.com/pepcid.html
http://www.rxmed.com/b.main/b2.pharmaceutical/b2.prescribe.html
http://www.rxlist.com/pepcid-drug.htm
http://gerd.emedtv.com/pepcid/pepcid-side-effects.html
http://medical-wiki.com/articles/pepcid-side-effects-pepcid-ac-side-effects/
概述
[edit | edit source]阿达拉特是拜耳公司生产的药物,其通用名为硝苯地平(Nifedipine)。它属于钙通道阻滞剂。其化学名为 3,5-吡啶二羧酸,1,4-二氢-2,6-二甲基-4-(2-硝基苯基)-二甲酯。该药物通常用于治疗高血压。该药物有片剂形式,口服后生物利用度为 84-89%。硝苯地平通过细胞色素 P450 3A4 系统代谢,该系统位于肠粘膜和肝脏中,其半衰期为 2 小时。该药物是一种 1,4-二氢吡啶类钙通道阻滞剂,是一种选择性血管扩张剂。通常情况下,在心脏中,钙离子与心脏内钙通道的结合会增加心率。心率的增加会通过收缩动脉壁来导致动脉收缩。该药物的作用实际上是阻止钙与心脏内的钙通道相互作用,这使得动脉壁扩张,从而防止高血压,同时减缓心率。一些严重的副作用包括低血压、心律不齐和充血性心力衰竭。与该药物一起食用葡萄柚也很重要。
副作用
[edit | edit source]服用阿达拉特后,如果您出现以下一种或多种症状,请联系医生。
- 荨麻疹
- 呼吸困难
- 嘴唇、面部、喉咙或舌头肿胀
- 心绞痛加重
- 感觉要晕倒
- 呼吸急促
- 四肢肿胀
- 心跳过快,怦怦直跳
- 麻木
- 黄疸(皮肤或眼睛发黄)
- 胸痛或压迫感
较轻微的副作用包括
- 头痛
- 眩晕
- 嗜睡
- 便秘
- 恶心
- 腹泻
- 胃痛
- 失眠
- 皮疹或瘙痒
- 关节疼痛
- 比平时排尿更多
与其他药物的不相容性
[edit | edit source]阿达拉特与以下一种或多种药物混合使用可能会产生严重的反应。
- 阿卡波糖
- 西咪替丁
- 芬太尼或其他麻醉性止痛药
- 地高辛
- 奈法唑酮
- 圣约翰草
- 利福布汀
- 血液稀释剂
- 抗真菌药物
- β-受体阻滞剂
- 心脏节律药物
- 艾滋病毒/艾滋病药物
- 抗癫痫药物
参考文献
[edit | edit source]http://www.drugs.com/adalat.html
http://www.medterms.com/script/main/art.asp?articlekey=3846
http://www.rxlist.com/adalat-drug.htm
http://online.factsandcomparisons.com

Bosulif(博舒替尼)是一种新药,最近被美国食品药品监督管理局(FDA)批准用于治疗费城染色体阳性的患者,费城染色体是一种基因突变,导致骨髓产生酪氨酸激酶。这种药物用于治疗慢性粒细胞白血病(CML)阳性患者。CML 阳性是指患者出现慢性期或加速期的 CML。Bosulif 是一种 Abl 和 Src 激酶抑制剂。它主要含有博舒替尼,如图所示。由于它是一种新药,建议仅在患者对其他治疗方法不能耐受时使用。随着这一批准,我们见证了这种治疗方法的有效改善。
对于对博舒替尼或该药物任何成分过敏的患者,不能使用 Bosulif。它以片剂形式生产,可以方便地与食物一起使用。避免与该药物一起食用柚子或葡萄汁,因为 Bosulif 在胃酸存在的情况下会被吸收,这意味着 Bosulif 的量会增加。您需要咨询您的医生以确定剂量,并且只服用医生开的剂量。如果使用不当,该药物也会有一些严重的副作用。
- 胃部问题、腹泻、呕吐、头痛、咳嗽、发烧。
- 血细胞计数低、肝脏问题。
- 呼吸急促、胸痛、体重增加。
http://www.drugs.com/bosulif.html http://www.medicalnewstoday.com/articles/249971.php
Luvox 是一种选择性5-羟色胺再摄取抑制剂(SSRI),通用名为氟伏沙明马来酸盐,化学名为 5-甲氧基-4'-(三氟甲基)戊酰苯酮-(E)-O-(2-氨基乙基)肟马来酸盐 (1:1)。它可用于治疗慢性抑郁症、强迫症 (OCD) 和社交焦虑症。该药物通过增加大脑中 5-羟色胺的含量来防止抑郁、焦虑和恐惧感。5-羟色胺是人体自然产生的神经递质,它可以维持精神平衡,并产生平静或快乐的感觉。[8]
该药物以片剂形式存在,在 20 到 2000 ng/mL 的浓度范围内,约 80% 的氟伏沙明与血浆蛋白结合,主要为白蛋白。氟伏沙明马来酸盐的绝对生物利用度为 53%。该片剂应口服,不要压碎或咀嚼。剂量通常由医生开具,从低剂量开始,随着时间的推移逐渐增加剂量。该药物的全部效果可能需要几周才能开始显现。
氟伏沙明马来酸盐在肝脏中被广泛代谢。主要的人体代谢产物是氟伏沙明酸,该药物在体内的半衰期为 15.6 小时。该药物靶向大脑中的化学失衡,有助于治疗慢性抑郁症。
在突触和神经细胞之间,存在神经递质的交换。5-羟色胺是这些神经递质之一,可以传递神经细胞之间的信息。神经细胞含有再摄取通道,可以在神经递质完成任务并不再需要时将其重新吸收。当信息交换更快时,5-羟色胺在突触中停留的时间更短,因为它被更快地重新吸收,据信这会导致抑郁症。如果体内的 5-羟色胺水平失衡,可能会出现强迫症或其他焦虑症。SSRI 类药物,如 Luvox,可以选择性地与 5-羟色胺的再摄取通道结合,从而阻止再摄取通道重新吸收 5-羟色胺。这会导致 5-羟色胺停留在突触中,从而有助于抵消抑郁感。研究表明,Luvox 是一种有效的药物,可以治疗成人和儿童的强迫症。[9]
一些严重的副作用包括癫痫发作、发烧和自杀念头增加。一些更严重的副作用包括胸痛、协调障碍、头晕、幻觉、疼痛、意识丧失、呼吸困难以及呕吐或排血。
服用此药物时,可能会遇到以下过敏反应的其他迹象。
- 皮肤疹或荨麻疹
- 呼吸困难
- 面部、嘴唇、舌头或喉咙肿胀
如果出现任何新的或加重的症状,例如情绪或行为改变、焦虑、睡眠障碍、惊恐发作或感觉冲动,应立即致电医生。如果您突然停止服用氟伏沙明,可能会出现一些戒断症状,包括易怒、头晕、头痛、情绪改变、疼痛、睡眠困难或四肢麻木。在您停止服用药物之前,医生应帮助您逐渐减少剂量。[10]

服用氟伏沙明之前,如果可能发生任何过敏反应,务必告知医生或药剂师,因为这种药物含有非活性成分,可能会导致过敏反应或其他严重问题。服用此类药物时要谨慎。请务必告知医生或药剂师您的病史,尤其是家族史疾病、个人史疾病等。
服用此药时,请记住,驾驶可能很危险,因为可能会出现头晕。因此,请勿操作机器或驾驶。此外,请勿进行任何需要高度警觉的活动,直到您确定进行该活动是安全的。请记住,服用氟伏沙明时,永远不要饮酒。
不同年龄的人可能会出现该药物的不同副作用。例如,老年人可能对副作用更敏感,尤其是出血副作用。服用利尿剂的老年人可能会在体内出现某种矿物质失衡。与老年人一样,儿童等年轻人也可能对该药物的副作用更敏感。儿童可能会出现食欲不振和体重减轻。因此,父母在服用此药物时,务必监测孩子的体重和身高。
服用氟伏沙明的孕妇可能会伤害她们腹中的胎儿。因此,最好在实际服用之前咨询医生。如果母亲注意到他们三个月的婴儿出现以下任何症状,应立即联系婴儿医生。
- 进食/呼吸困难
- 癫痫发作
- 肌肉僵硬
- 持续哭泣
母乳喂养的母亲应咨询医生。
- ↑ http://www.chemistryexplained.com/A-Ar/Acetaminophen.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmed/20020268
- ↑ http://tuftsjournal.tufts.edu/2008/04/professor/01/
- ↑ http://dmd.aspetjournals.org/content/31/12/1499.full
- ↑ http://www.tylenolprofessional.com/pharmacology.html
- ↑ http://cid.oxfordjournals.org/content/31/Supplement_5/S211.full
- ↑ http://news.consumerreports.org/health/2011/11/repeatedly-taking-too-much-tylenol-can-be-dangerous.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000955/
- ↑ http://anxiety.emedtv.com/luvox/luvox-p2.html
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000955/
http://www.medicinenet.com/fluvoxamine/article.htm
http://online.factsandcomparisons.com
炔诺酮 (或诺雷思特龙) 是一种女性激素,可以阻止排卵(卵子从卵巢中释放)。这种药物会改变女性的宫颈粘液和子宫内膜,使精子更难到达子宫,从而更难受孕。
炔诺酮用于避孕 (避孕) 以预防怀孕。炔诺酮还用于治疗月经失调、子宫内膜异位症或由激素失衡引起的异常阴道出血。
如果您在服用诺雷孕酮后出现以下任何过敏反应迹象,请立即寻求医疗帮助
- 荨麻疹
- 呼吸困难
- 脸部、嘴唇、舌头或喉咙肿胀。
如果您出现以下任何严重副作用,请停止使用此药物并致电您的医生
- 突然麻木或无力,尤其是身体一侧;
- 突然头痛、意识混乱、眼后疼痛、视力、言语或平衡问题;
- 一侧或两侧腿部疼痛或肿胀;
- 偏头痛;
- 手或脚肿胀,体重迅速增加;
- 抑郁症状(睡眠问题、无力、情绪变化);
- 严重的盆腔疼痛;
- 胸痛或沉重感,疼痛蔓延到手臂或肩膀,恶心,出汗,全身不适;或
- 恶心、胃痛、低烧、食欲不振、深色尿液、陶土色便、黄疸(皮肤或眼睛发黄)。
- 较轻微的副作用可能包括
- 轻微恶心、呕吐、腹胀、胃痉挛;
- 乳房疼痛、肿胀或压痛;
- 头晕;
- 雀斑或面部皮肤变黑;
- 痤疮或毛发生长增加;
- 体重变化;
- 阴道瘙痒或分泌物;
- 皮肤瘙痒或皮疹;
- 月经周期变化,性欲下降;或
- 轻微头痛。
1951 年,诺雷孕酮由化学家 Luis Miramontes、Carl Dijerassi 和 George Rosenkranz 在墨西哥城的 Syntex 公司合成。
19-去甲-17α-乙炔睾酮的合成


官能团:酮、烯烃、甲基(2)、醇、炔烃
- http://www.drugs.com/mtm/norethindrone.html
- Djerassi, C.; Miramontes, L.; Rosenkranz, G.; Sondheimer, Franz (1954)。“类固醇。LIV。19-去甲-17α-乙炔睾酮和 19-去甲-17α-甲基睾酮的合成”。J. Am. Chem. Soc. 76 (16): 4092–94. doi:10.1021/ja01645a010
- http://commons.wikimedia.org/w/index.php?title=Special%3ASearch&profile=default&search=Norethisterone+&fulltext=Search
紫杉醇 (Taxol) 是一种重要的抗癌药物。1967 年,Monreoe E. Wall 和 Mansukh C. Wani 从太平洋紫杉树 Taxus brevifolia 的树皮中提取了紫杉醇。
紫杉醇用于治疗英国的艾滋病相关卡波西肉瘤、乳腺癌、卵巢癌和肺癌。这种药物与微管蛋白结合并抑制微管解聚。这使微管稳定并阻止进一步的细胞分裂。
如果您出现以下副作用,请寻求医疗救助
- 荨麻疹
- 呼吸困难
- 感觉自己可能昏倒
- 面部、嘴唇、舌头或喉咙肿胀
- 心率缓慢
- 癫痫发作
- 皮肤苍白,容易瘀伤或出血,异常虚弱
- 发烧、发冷、身体疼痛、流感症状
- 嘴巴或嘴唇内出现溃疡
- 麻木
- 血压升高
- 关节和肌肉疼痛
- 轻微恶心和呕吐
- 脱发


(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-二乙酰氧基-15-{[(2R,3S)-3-(苯甲酰氨基)-2-羟基-3-苯基丙酰基]氧基}-1,9-二羟基-10,14,17,17-四甲基-11-氧代-6-氧杂四环[11.3.1.0~3,10~.0~4,7~]十七碳-13-烯-2-基相对苯甲酸酯的结构
官能团:芳香族 (3)、酰胺 (1)、酮 (2)、酯 (4)、醇 (3) 和醚 (5) 它有 11 个立体中心。

微管是细胞成分,充当细胞骨架。为了发生细胞分裂,微管需要解聚回微管蛋白。在那之后,微管蛋白重新聚合形成细胞分裂纺锤体。复制染色体在有丝分裂过程中的移动取决于纺锤体,因此也取决于微管的解聚。
紫杉醇或 Taxol 增强了微管蛋白聚合成稳定的微管,并且还直接与微管相互作用,使其稳定以防止解聚。因此,它干扰了纺锤体形成过程。染色体无法移动到分裂细胞的相对两侧。细胞分裂受到抑制,最终诱导细胞死亡。
Borad, Brijesh。 “与单独使用紫杉醇相比,紫杉醇和维生素 E 琥珀酸酯联合对 MCF-7 乳腺癌细胞培养的细胞毒性作用,通过 MTT 测定法。”PharmaTutor。N.p.. 网页。2012 年 12 月 7 日。
- http://www.rxlist.com/taxol-drug.htm
- http://www.cancer.gov/cancertopics/druginfo/paclitaxel
- http://en.wikipedia.org/wiki/Paclitaxel#Mechanism_of_action
Allegra 的通用名称为 Fexofenadine,属于组胺 H1 受体拮抗剂。它由 Advancis Pharmaceutical Corporation 制造,化学名称为 (±)-4-[1 羟基-4-[4-(羟基二苯甲基)-1-哌啶基]-丁基]-,-二甲基苯乙酸盐酸盐。主要用途之一是治疗过敏引起的症状。盐酸非索非那定 60% 至 70% 与血浆蛋白结合,主要是白蛋白和 1-酸性糖蛋白。大约 5% 的非索非那定盐酸盐总剂量通过肝脏代谢通过代谢物特非那定消除。60 毫克的半衰期为 14.4 小时。药物通过竞争性抑制 H-1 受体位点并防止组胺-1 附着在受体位点以引起过敏症状而起作用。组胺-1 在存在身体过敏的某些刺激物时由肥大细胞释放。该药物不会阻止肥大细胞释放组胺-1,而是与组胺-1 底物竞争受体位点的位置。

非索非那定用于缓解季节性过敏性鼻炎(如花粉症)的过敏症状。2 岁以上儿童和成人的一些过敏症状包括流鼻涕;打喷嚏;眼睛发红、发痒或流泪;或鼻子、喉咙或口腔上颚发痒。它还用于缓解荨麻疹的症状,如皮肤上出现红色、发痒的隆起区域,包括 6 个月以上儿童和成人的瘙痒和皮疹。总的来说,非索非那定属于一类称为抗组胺药的药物。抗组胺药能阻断组胺的作用,组胺是体内引起过敏的物质。非索非那定仅用于控制季节性过敏性鼻炎和荨麻疹的症状,但它不能治愈这些疾病。
非索非那定以片剂或液体形式口服。服用该药物的人通常每天服用一次或两次,具体取决于过敏症状的严重程度。不要服用任何果汁,如橙汁、葡萄柚汁或苹果汁,因为非索非那定在人体系统中的作用不会那么有效。请记住,每天在同一时间左右服用此药。仔细阅读处方标签上的说明,并按要求服用。不要服用超过或少于规定的剂量,也不要比医生规定的次数更频繁地服用。如果服用非索非那定液体,使用前务必充分摇匀瓶子。
副作用
[edit | edit source]服用非索非那定可能会引起某些副作用。如果以下任何副作用严重且没有消失,务必尽快咨询医生。
- 头痛
- 眩晕
- 腹泻
- 呕吐
- 手臂、腿部或背部疼痛
- 月经期间疼痛
- 咳嗽
注意:出现严重副作用,应立即致电医生,包括荨麻疹、皮疹、瘙痒、呼吸或吞咽困难以及面部或嘴唇肿胀。
合成
[edit | edit source]非索非那定可以通过从哌啶-4-羧酸酯和4-溴苯基乙腈合成,如下所示。[1][2]

参考文献
[edit | edit source]http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001008/
- ↑ Daniel Lednicer (1999). The Organic Chemistry of Drug Synthesis. Vol. 6. New York: Wiley Interscience. ISBN 0-471-24510-0.
- ↑ Kawai, SH; Hambalek, RJ; Just, G (1994). "A facile synthesis of an oxidation product of terfenadine". J. Org. Chem. 59 (9): 2620–22. doi:10.1021/jo00088a056.
{{cite journal}}: Unknown parameter|month=ignored (help)
http://www.medicinenet.com/fexofenadine-oral/article.html

简介
[edit | edit source]阿德拉尔是一种特定的品牌药物,主要用于治疗多动症(注意力缺陷多动障碍),但也可以用于治疗某些类型的嗜睡症。右旋苯丙胺-苯丙胺,也称为阿德拉尔,是一种处方药,可以帮助患有注意力障碍(如注意力缺陷多动障碍 (ADHD))和睡眠障碍(如嗜睡症)的患者集中注意力。阿德拉尔在美国属于 II 类药物,这意味着它有导致生理和心理依赖的可能性,有很高的滥用潜力(因为它是由四种苯丙胺盐混合而成),但已被批准用于医疗用途 [1]。阿德拉尔是一种兴奋剂,这意味着它是一种可以改善心理和身体机能的精神活性药物。它也是苯乙胺和苯丙胺家族的近亲。</g>

医疗用途
[edit | edit source]如上所述,阿德拉尔是一种处方药,用于治疗多种类型的 ADHD(比同龄人更容易出现注意力集中、控制行为、保持安静或不动)和嗜睡症(一种睡眠障碍,会导致白天过度嗜睡和突发的睡眠发作)[1],尽管它的主要作用是治疗 ADHD 患者。市场上常见的两种形式是:阿德拉尔 XR(缓释)和阿德拉尔 IR(速释)。两者均用于治疗 ADHD,但只有 IR 形式用于治疗嗜睡症。阿德拉尔被认为可以阻止诸如去甲肾上腺素和多巴胺等化学物质的再摄取,这些化学物质被认为可以更好地帮助 ADHD 患者,因为这些化学物质的不同水平应该会导致更好的注意力集中能力、行为控制能力以及最终在同龄人中保持安静或不动。
副作用
[edit | edit source]许多副作用包括:紧张、烦躁不安、入睡困难或睡眠中断、身体部位无法控制的颤抖、头痛、性欲或性能力改变、口干、胃痛、恶心、呕吐、腹泻、食欲不振、体重减轻。也可能出现其他更严重的不良反应。
化学
[edit | edit source]

如前所述,阿德拉尔属于苯乙胺和苯丙胺家族。苯乙胺是一种有机化合物,是许多具有精神活性和兴奋作用的药物的一部分 [2],通常存在于兴奋剂、迷幻药、致幻剂、食欲抑制剂、支气管扩张剂、鼻腔充血剂和抗抑郁剂中 [3]。苯丙胺是精神兴奋剂,是苯乙胺类的一种分支,它可以促进注意力集中和清醒。更具体地说,右旋苯丙胺是苯丙胺的立体异构体。前缀“右旋-”表示偏振光平面顺时针旋转。由于阿德拉尔由右旋苯丙胺和苯丙胺组成;它由苯丙胺的两种对映异构体组成。
生化相互作用/组成
[edit | edit source]阿德拉尔是一种苯丙胺,可以描述为具有中枢神经系统兴奋作用的非儿茶酚胺拟交感神经胺。本质上,它是一种苯丙胺盐的混合物,被认为可以阻止去甲肾上腺素和多巴胺再摄取回神经元。在阿德拉尔 XR 形式中,药物的组成包括 d-苯丙胺和 l-苯丙胺,比例为 3:1。阿德拉尔 XR 的四种成分包括右旋苯丙胺酒石酸盐、苯丙胺天冬氨酸盐、右旋苯丙胺硫酸盐和苯丙胺硫酸盐,每种成分的比例相同。
药理学
[edit | edit source]右旋苯丙胺 (d-AMP) 在治疗 ADHD 中的具体机制尚不清楚,但已知它在多巴胺神经传递的效率中起着重要作用。右旋苯丙胺的立体异构体苯丙胺被认为通过使用多巴胺活性转运体 (DAT) 作为载体从神经末梢释放多巴胺 [4]。尽管阿德拉尔用于增强注意力集中,但研究表明,苯丙胺不能优化短期记忆,也不能改善“认知灵活性”。事实上,有证据表明,它可能损害短期记忆和认知灵活性 [5]。d-AMP 也被认为是再摄取抑制剂,因为它是一种底物类似物,因此它与具有类似单胺基团的神经递质(儿茶酚胺)如多巴胺竞争摄取 [6]。由于 d-AMP 通过提高多巴胺浓度来诱导多巴胺效应,使用者会感受到兴奋性神经递质的影响。在极高剂量下,d-AMP 会通过触发儿茶酚胺级联反应,释放大量神经递质和过度刺激受体,从而导致欣快感 [8]。据推测,d-AMP 倾向于与多巴胺系统相互作用,而它的对映异构体倾向于与去甲肾上腺素系统相互作用 [9]。
Silverston 等人(2002 年)在双相情感障碍方面的研究推测了右旋苯丙胺化学上影响人体的另一种方式。研究表明,右旋苯丙胺通过多巴胺和去甲肾上腺素提高磷脂酰肌醇循环的活性。磷脂酰肌醇循环由 Lowell 和 Hopkins 发现,已知它可以产生磷脂酰肌醇,磷脂酰肌醇是真核生物中脂类信号分子的前体,有助于调节特定功能 [10]。
药代动力学
[edit | edit source]根据阿德拉尔 XR® 的药物说明书,成人 d-AMP 的平均消除半衰期为 10 小时;体重小于或等于 165 磅的个体为 11 小时,6-12 岁儿童为 9 小时。在 l-苯丙胺的对映异构体中,平均消除半衰期为 13 小时,体重小于或等于 165 磅的个体为 13-14 小时,6-12 岁儿童为 11 小时 [11]。
代谢
[edit | edit source]尚未确定所有代谢苯丙胺的酶。然而,CYP2D6 负责生成 4-羟基苯丙胺,该物质进而氧化为 α-羟基苯丙胺,并经过进一步的未知代谢。已知苯丙胺也是 CYP2D6 酶的抑制剂 [11]。
苯丙胺对 pH 值敏感,pKa 为 9.9,因此其妥善处置依赖于尿液的 pH 值。“酸性 pH 值和高流量导致肾脏消除增加……表明存在主动分泌作用 [11]。”
阿得拉被滥用为
- 学习药:其绰号包括“大学毒品”或“认知类固醇”。服用阿得拉用于学习的学生报告说他们能够很好地集中精力于书本,并且可以在没有药物的情况下比没有药物的情况下在随后的考试中取得更好的成绩。 - 派对药物:阿得拉含有右旋苯丙胺,它能带来幸福感、自信和增强性欲的感觉,并能让使用者长时间不睡觉。 - 减肥药:抑制食欲通常是阿得拉的副作用,并且深受想要轻松减肥的年轻女性的喜爱。
为什么阿得拉会被滥用?阿得拉在治疗许多 ADHD 病例方面具有相同的特性,这些特性使其成为非 ADHD 学生的首选药物,他们发现阿得拉对他们有其他吸引力。阿得拉通过改变大脑的化学成分来起作用,从而帮助 ADHD 患者尽管受到干扰,也能专注于手头的任务。对于非 ADHD 患者,阿得拉会导致专注力和能量的增加,以及相关的食欲下降。虽然在许多学校中,学生之间偶然服用阿得拉被认为是可以接受的,但为了非处方目的或由未被处方的人服用任何处方药,都构成滥用。
滥用阿得拉的后果
误用或滥用阿得拉会导致一系列问题,包括难以入睡、敌意感、焦虑或偏执,以及过度活跃、兴奋的状态。有些使用者可能会出现不希望出现的或不健康的体重减轻。特别是鼻吸时,阿得拉会导致心脏率、体温以及血压的潜在危险性升高。护理人员会对 ADHD 患者进行筛查,以避免将阿得拉开给那些服用该药会使其处于危险境地的人。没有处方就服用该药的滥用者,无法享受这种筛查的好处。具有讽刺意味的是,尽管阿得拉在校园里被滥用的主要原因是提高学业成绩,但服用该药会导致狂躁状态的学生可能无法对他们在阿得拉的影响下产生的论文或考试质量做出良好的判断。滥用阿得拉后,使用者通常会出现“崩溃”并遭受疲劳、恶心、抑郁或烦躁。有些阿得拉滥用者吸食大麻作为这些令人苦恼症状的解药。长期滥用后,你的身体会依赖阿得拉才能正常运作,试图停止服用该药会导致戒断症状,包括恐慌、自杀念头和噩梦。
http://adhd.emedtv.com/adderall/adderall-abuse.html http://adderallabuse.com/ http://www.webmd.com/drugs/drug-63163-Adderall+Oral.aspx?drugid=63163&drugname=Adderall+Oral http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000166/ http://www.drugs.com/pro/adderall.html 1. 21 USC § 812 – Schedules of controlled substances | LII / Legal Information Institute http://www.law.cornell.edu/uscode/text/21/812
2. Glen R. Hanson, Peter J. Venturelli, Annette E. Fleckenstein (2005-11-03). "Drugs and society (Ninth Edition)". Jones and Bartlett Publishers. ISBN 978-0-7637-3732-0
3. 苯乙胺. http://en.wikipedia.org/wiki/Phenethylamine
4. Kuczenski, R., and D. S. Segal. 1997. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J. Neurochem. 68:2032–2037.
5. Lackhan S. et al. (2012 年 6 月 15 日). "Attention deficit hyperactivity disorder 有无个体中的处方兴奋剂:滥用、认知影响和不良反应". 脑与行为。
6. Kuczenski R 等人。(1995 年 2 月 1 日)。“海马体去甲肾上腺素、尾状核多巴胺和血清素以及对苯丙胺和甲基苯丙胺立体异构体的行为反应”。神经科学杂志 15(2):1308-1317。
7. Patrick 和 Markowitz;Markowitz, John S. (1997). "Pharmacology of Methylphenidate, Amphetamine Enantiomers and Pemoline in Attention-Deficit Hyperactivty Disorder". 人类精神药理学 12(6):527-546(页:530)。
8. InterScience | 右旋苯丙胺增加志愿者的磷酸肌醇循环活性:一项 MRS 研究
9. http://en.wikipedia.org/wiki/Adderall
10. Heck, J. et.al, 'A Conspicuous Connection: Structure Defines Function for the Phosphatidylinositol-Phosphate Kinase Family', Critical Reviews in Biochemistry and Molecular Biology, 42:1, 15 – 39
11. FDA (2007)。阿得拉 XR 药物指南。第 1-14 页。存档于 2011 年 1 月 30 日。
利他能,也称为哌甲酯,是一种作用于中枢神经系统的精神兴奋剂药物。自 1960 年以来,它通常被处方用于治疗 ADHD(或注意力缺陷多动障碍)和嗜睡症患者;然而,它在 1990 年代广泛传播,当时 ADHD 得到了更广泛的认可。哌甲酯是一种白色、无味、细小的结晶粉末,呈酸性。利他能易溶于水和甲醇,溶于酒精,微溶于氯仿和丙酮。
历史
哌甲酯于 1944 年首次合成,并在 1954 年被鉴定为兴奋剂。哌甲酯由 Ciba(现为诺华)化学家 Leandro Panizzon 合成。他的妻子 Marguerite 血压低,在打网球之前会服用这种药作为兴奋剂。他以他妻子的昵称 Rita 将这种物质命名为利他能。最初,它被销售为两种外消旋体的混合物,80% (±)-赤型和 20% (±)-苏型。随后对外消旋体的研究表明,中枢兴奋作用与苏型外消旋体相关,并专注于赤型异构体分离成活性更高的苏型异构体。从 1960 年代开始,它被用于治疗患有 ADHD 或 ADD 的儿童,当时被称为多动症或轻度脑功能障碍 (MBD)。哌甲酯的生产和处方在 1990 年代显著增加,特别是在美国,因为 ADHD 的诊断在医疗和心理健康领域得到了更深入的理解和更普遍的认可。2000 年,杨森获得美国食品药品监督管理局 (FDA) 批准,将其销售为“专注达”。有关专注达的更多信息,请参阅本文的“缓释”部分。
哌甲酯有副作用;如果这些副作用严重或没有消失,必须联系医生
- 紧张
- 难以入睡或保持睡眠
- 头晕
- 呕吐
- 食欲不振
- 胃痛
- 腹泻
- 恶心
- 烧心
- 口干
- 头痛
- 肌肉紧张
- 身体某一部分不受控制地运动
- 烦躁不安
- 四肢麻木、灼痛或刺痛
- 性欲下降
- 痛经
利他能有不同的形式,包括:速释片、咀嚼片、液体、缓释片、缓释胶囊和缓释片。
所有形式的哌甲酯均口服。成年人通常每天服用三次该药物,而儿童通常每天服用两次,通常在饭前服用。为避免无法入睡的副作用,患者应在晚上 6 点后不要再服用最后一次剂量。
http://www.rxlist.com/ritalin-drug.htm
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000606/
http://psychopharmacopeia.com/index.php?generic=methylphenidate
帕罗西汀(也称为舍瑞普汀、思瑞特、阿罗帕克斯和帕罗西汀)是一种抗抑郁药,属于一类称为选择性血清素再摄取抑制剂 (SSRI) 的分子。它可用于治疗抑郁症、强迫症、惊恐症、社交焦虑症、创伤后应激障碍和经前期焦虑症。帕罗西汀是一种口服精神药物,以片剂形式存在,其分子式为 C19H20FNO3•HCl•1/2H2O,分子量为 374.8。
帕罗西汀用于治疗抑郁症,每天早上服用一次,每次 20 毫克。剂量范围可能在 10 毫克到 50 毫克之间,应每周复查。该药物已知在服用一年内有效。帕罗西汀片剂存在不同的浓度,由颜色标识:10 毫克 - 黄色,20 毫克 - 粉色,30 毫克 - 蓝色,40 毫克 - 绿色。应储存在 25°C 或更低的温度下。该药物已被证实对孕妇有致畸作用,因此在服用前应予以考虑。研究发现,服用帕罗西汀的母亲所生的新生儿会出现肺部和心脏疾病。美国食品药品监督管理局将其列为黑盒警告,不建议 18 岁以下的未成年人使用。帕罗西汀不应与 Orap、Mellaril、Zyvox、Urolene Blue、MAOI、Furoxone、Marlplan、Nardil、Azilect、Eldepryl 或 Parnat 合用,以避免不良药物相互作用。此外,还应告知医生任何肝脏或肾脏疾病、血液凝固障碍、抑郁症史和自杀念头史。
如果您出现以下任何副作用,请联系您的医生。
- 骨痛、肿胀、压痛
- 激动、幻觉
- 肌肉僵硬
- 头痛
- 性欲下降
- 轻度恶心、便秘
- 体重变化
- 口干、打哈欠、耳鸣
- 自杀行为
"家庭药物 A-z 列表帕罗西汀(盐酸帕罗西汀)副作用药物中心帕罗西汀(盐酸帕罗西汀)药物 - 药物指南和患者信息。"帕罗西汀(盐酸帕罗西汀)药物信息:药物指南和患者信息。 N.p.,n.d. 网页。 2012 年 12 月 7 日。
"帕罗西汀。"维基百科。维基媒体基金会,2012 年 6 月 12 日。网页。 2012 年 12 月 7 日。
"帕罗西汀。"来自 Drugs.com 的信息。 N.p.,n.d. 网页。 2012 年 12 月 7 日。
安定,通用名叫做甲丙氨酯,是一种镇静剂,用于治疗焦虑和短期控制失眠。它是一种氨基甲酸酯衍生物,曾经用作轻度镇静剂。
甲丙氨酯于 1950 年 5 月由 Bernard John Ludwig 博士和 Frank Milan Berger 博士在卡特产品公司首次合成。它于 1955 年上市,迅速成为美国历史上第一种畅销的精神药物,在好莱坞流行起来,并因其看似神奇的效果而声名大噪。
在 20 世纪 40 年代中期,Berger 博士在一家英国制药公司的实验室工作,寻找青霉素的防腐剂,当时他注意到一种叫做甲苯二酚的化合物在小型实验室动物中具有镇静作用。然而,甲苯二酚作为镇静剂的使用有三个主要缺点:作用持续时间很短,对脊髓的影响大于对大脑的影响,以及活性较弱。Berger 博士搬到新泽西州的华莱士实验室后,他与化学家 Bernard Ludwig 博士一起合成了一种与之相关的镇静化合物甲丙氨酯,该化合物能够克服这三个缺点,并开始销售这种新药。
尽管甲丙氨酯被标记为更安全,但它与巴比妥类药物具有许多药理作用和危险性。
甲丙氨酯的作用机制尚不清楚,但研究表明它会影响中枢神经系统的多个部位,包括边缘系统和丘脑系统。甲丙氨酯与 GABAA 受体结合,中断网状结构和脊髓的神经传递,导致镇静和疼痛感知改变。
过量服用会引起昏迷、嗜睡、肌肉控制丧失、呼吸困难、迟钝和无反应等症状。据报道,摄入仅 12 克即可导致死亡,而摄入多达 40 克则可以存活。
甲丙氨酯,2-甲基-2-丙基-1,3-丙二醇二氨基甲酸酯,合成方法如下。2-甲基戊醛与两个甲醛分子反应,随后将得到的 2-甲基-2-丙基丙烷-1,3-二醇通过与光气和氨的连续反应转化为二氨基甲酸酯。

http://www.flexyx.com/P/Paxin.html 来自维基百科共享资源的图片

大麻是一种精神活性药物,也称为大麻或草。它被归类为镇静催眠药或迷幻药,会扭曲感知。
许多人发现了吸食大麻烟的证据。在世界不同国家发现了公元前 3 千年的大麻烟吸食证据。例如,这些种子是在现在的罗马尼亚的一个古代墓葬遗址中发现的。不仅在墓葬遗址中发现了大麻种子,而且在中国新疆维吾尔自治区西北部,在一名 2500-2800 年前的木乃伊萨满旁边,发现了一个装满大麻叶碎片和种子的皮革篮子。此外,几千年前印度和尼泊尔的许多古代印度教徒也使用大麻。因此,许多人使用大麻作为一种添加剂、治疗方法或享受方式。
John Gregory Bourke 也在 19 世纪后期描述了“mariguan”的使用。Bourke 将 mariguan 描述为大麻 indica。1894 年,德克萨斯州格兰德河地区许多墨西哥居民使用它。墨西哥居民通常将其用作治疗哮喘的来源,以加快分娩速度,驱除巫婆,甚至作为一种爱情药水。
此外,mariguan 也是许多人所知的一些被称为 loco weed 的植物之一。它也被比作大麻。然而,大麻会导致身体退化和痴呆的出现;因此,法律禁止出售它,因为它对人们的健康有害。
此外,从 20 世纪初开始,大麻在世界各国都被定为违禁品。例如,美国人从 1909 年开始被禁止销售大麻。它在 20 世纪也分别在南非、牙买加、英国和新西兰被禁止。在 1923 年的鸦片和毒品法案中,加拿大也将其定为违禁品。
1937 年,美国通过了《大麻税法》。这导致禁止生产和销售大麻和工业大麻。因此,任何被发现出售或使用大麻或工业大麻的人,都会因为违反《大麻税法》而被罚款。然而,在 2003 年,欧盟 95% 的大麻麻皮被用作动物垫料。欧盟的建筑工人也使用大麻麻皮作为建筑材料。
因此,从大麻的历史来看,可以说大麻可以是好的,也可以是坏的,这取决于它的使用方式。某些国家,例如通过了《大麻税法》的美国,将其用作减少大麻使用人数的来源,因为有人声称它对人体有害。
如果适度服用,大麻似乎几乎没有长期影响,而且上瘾的可能性很低。遗传因素可能会影响一个人对上瘾的易感性。虽然身体上瘾的可能性为零,但心理上瘾是可能的。
大麻是一种止痛药和镇静剂。大麻使用者经常描述吸食大麻的体验是放松和轻松的,产生一种朦胧和头晕目眩的感觉。使用者的眼睛可能会扩张,导致颜色看起来更加强烈,其他感官也可能会增强。后来,使用者可能会感到偏执和恐慌,尽管这种情况很少见。THC 与大脑的相互作用会导致这些感觉。它还可以减轻治疗癌症的药物引起的恶心。它可以降低青光眼患者眼内的压力,减轻某些运动障碍的症状,并干扰记忆。它会改变听觉感知,并刺激食欲。长期使用与短期记忆障碍相关。
大麻主要分为两种类型:Sativa 和 Indica。Indica 品种以体型较小而闻名,其特征是矮壮、深绿色、叶片茂密,同时花苞和小叶上覆盖着大量的树脂腺。这种品种的使用者会感到身体平静而宁静。这种品种经常被吸食者用来治疗失眠,同时为他们带来放松和缓解压力,这就是为什么 Indica 品种更适合在夜间使用的原因。另一方面,Sativa 品种则高大、纤细、浅绿色,叶子比 Indica 窄。尽管这两种类型都能使吸食者感到放松,但 Sativa 却以其活力和刺激作用而闻名,同时也能提高创造力和专注力。Sativa 的刺激作用使其成为白天使用的更好选择。两种品种的种植方式也有所不同。通常情况下,Indica 植株是在室内种植的,而 Sativa 植株则是在室外种植的,因为它们体型较大(Sativa 比 Indica 长得高)。此外,还种植了两种品种的组合,称为杂交品种,其比例范围从 30% Sativa - 70% Indica 品种、80% Sativa - 20% Indica 品种,以及许多 50% - 50% 的组合类型。
加工
Kief 是大麻的一种加工形式。Kief 是一种富含毛状体的粉末形式。它也可以从大麻植物的叶子和花朵中筛选出来。使用 Kief 的人要么直接使用粉末形式,要么将其压缩成大麻饼。
大麻实际上是女大麻植物的花朵中提取的浓缩树脂。因此,根据其纯度,它的颜色从金棕色到黑色不等。大麻甚至比纯大麻更有效。使用大麻的人要么吸食它,要么咀嚼它。
还有大麻油或“丁烷蜂蜜油”(BHO)。大麻油是用各种溶剂从成熟的大麻叶片中提取的,它是一种混合了有用的油脂和树脂的混合物。树脂也称为残留物,因为它的 THC 粘性强,通常具有粘性。因此,这种类型的油经常用在不同的含有大麻的食物中,因为它通常含有高比例的类大麻素,范围在 40-80% 之间。
未加工
许多人知道大麻或大麻是由女大麻植物的干燥、治愈的花朵、下部叶子和茎组成。这种形式是最常见的食用方式,它含有 3-28% 的 THC。


大麻中的活性成分是四氢大麻酚 (THC),它会刺激大脑中发现的某些类大麻素受体。人体中有两种类大麻素受体,即 CB1 和 CB2 受体。CB1 受体位于大脑、脊髓和周围神经系统区域,而 CB2 受体则位于免疫细胞中。类大麻素受体是 G 蛋白偶联受体 (GPCR) 超家族的成员,通常被认为介导腺苷酸环化酶活性的抑制,从而降低环状 AMP 水平。
这些受体位于某些神经元的终末按钮上,在正常情况下,它们调节神经递质的释放。类大麻素受体被一种称为神经酰胺的神经递质激活。神经酰胺属于一类称为类大麻素的化学物质。THC 是一种类大麻素化学物质,它模拟神经酰胺的作用,这意味着 THC 与类大麻素受体结合并激活神经元,从而对精神和身体造成影响。
当类大麻素受体被激活时,它们会打开终末按钮中的钾通道,缩短动作电位的持续时间和神经递质的释放。内源性大麻素,神经酰胺和 2-花生四烯酸甘油 (2-AG) 是 CB1 受体的两种天然配体。它们在饥饿、情绪、记忆和疼痛方面的正常功能。神经酰胺是在需要时产生和释放的,它不会储存在突触囊泡中。脂肪酰胺水解酶 (FAAH) 是一种抑制神经酰胺的酶。FAAH 还通过诱导酶促水解来使内源性大麻素在释放后失活。内源性大麻素的作用持续时间比 THC 短得多。THC 和内源性大麻素在阿片类药物的增强效应中发挥着重要作用。THC 会影响与记忆有关的区域,例如前额叶皮层、小脑、基底神经节、杏仁核和海马体。
海马体、小脑和基底神经节中存在高浓度的类大麻素受体。海马体位于颞叶内,对短期记忆很重要。当 THC 与海马体内的类大麻素受体结合时,它会干扰对近期事件的回忆。THC 还会影响由小脑控制的协调性。基底神经节控制着无意识的肌肉运动,这也是在服用大麻影响下运动协调性受损的另一个原因。
虽然大麻的药用使用被认为是安全的,只要正确服用作为处方药,但它也存在一些缺点,例如吸食和通过肺部吸入会导致某些副作用。它可能通过其引起心率加快和血压升高的作用导致心脏病。此外,它可能削弱一个人的免疫系统,从而使一个人的身体更容易受到感染和疾病的侵害。在怀孕期间,吸入 THC 会削弱胎儿的生长,同时还有可能将 dronabinol (THC) 注射到母乳中。因此,大麻与儿童白血病有关(有争议)。最后,当大麻用于治疗抑郁症以控制戒断症状时,它可能会适得其反,当一个人无法获得这种药物时会引发更多抑郁症。它会产生精神上的影响,导致更高的成瘾和强迫症风险。
虽然大麻一直被认为是一种娱乐性药物,但它也广泛用作一种医疗治疗形式。医用大麻可以用来治疗癌症、青光眼、艾滋病/艾滋病毒、多发性硬化症和其他疾病的症状。它还被发现可以缓解某些痉挛性和运动障碍的症状。大麻能够减轻患者的症状,这得益于其化学成分。称为类大麻素的化合物会激活体内的类大麻素受体,并能刺激食欲、减轻恶心和呕吐、缓解疼痛和抑制肌肉痉挛。
许多医生建议癌症患者使用医用大麻来帮助减轻治疗带来的疼痛症状并增加食欲。医用大麻被发现可以增加食欲,减轻化疗和放疗患者的疼痛。它还可以减轻恶心和呕吐的感觉。加州太平洋医疗中心研究所进行的研究表明,大麻二酚 (CBD) 可能通过阻断 Id-1 基因的活性来帮助阻止乳腺癌的扩散,该基因被认为会导致癌细胞的积极扩散到原始肿瘤部位以外的其他部位,也被称为转移。在实验室进行的其他研究表明,THC 和 CBD 会阻碍某些在实验室培养皿中生长的癌细胞的生长,甚至可能导致这些癌细胞死亡。还有一些动物研究也显示了相同的结果。然而,尚未对人体进行测试以确定 THC 和其他类大麻素是否降低了患癌风险。
有一些研究表明吸食大麻可能对肺部有害,甚至可能引发某些类型的癌症(有争议)。医用大麻药房现在正在推广食用产品,这些产品是含有 THC 提取物混合在黄油中的食物,供持有医用大麻许可证的患者食用。患者可以选择食用含有大麻的食物,而不必吸食大麻。食用产品的效果比吸入的效果持续时间更长。
"大麻的公认药用:基础研究。"http://www.drugscience.org/amu/amu_basic_research.html 2002 年重新安排大麻(大麻)的请愿书。Drug Science.org,2006 年。网络。2012 年 10 月 26 日。
Bonsor, Kevin. "大麻如何起作用" 2001 年 7 月 2 日。HowStuffWorks.com。<http://health.howstuffworks.com/wellness/drugs-alcohol/marijuana.htm> 2010 年 11 月 21 日。
biopsychiatry.com。 "THC 如何影响大脑。"http://www.a1b2c3.com/drugs/brn005.htm www.a1b2c3.com。2010 年 10 月 15 日。 . 2010 年 10 月 15 日。
"大麻二酚。"Cacycle。2010 年 3 月 5 日。网络。2012 年 10 月 26 日。
“大麻。”维基百科,自由的百科全书。维基媒体基金会,股份有限公司。2010 年 5 月 20 日。网络。2010 年 5 月 22 日。
Carlson, Neil R. 行为生理学。波士顿:皮尔森教育,股份有限公司,2007 年。
“内源性大麻素系统。”维基百科,自由的百科全书。维基媒体基金会,股份有限公司。2010 年 4 月 21 日。网络。2010 年 5 月 23 日。
http://en.wikipedia.org/wiki/Medical_cannabis
http://en.wikipedia.org/wiki/Effects_of_cannabis
King, Jason. 大麻圣经。中国:10 速出版社,2001 年。1-25。打印。
"大麻。"大麻。美国癌症协会,2012 年 7 月 13 日。网络。2012 年 10 月 26 日。<http://www.cancer.org/treatment/treatmentsandsideeffects/complementaryandalternativemedicine/herbsvitaminsandminerals/marijuana>。
"THC。"维基百科,自由的百科全书。维基媒体基金会,股份有限公司。2009 年 9 月 1 日。网络。2012 年 10 月 26 日。
"大麻。"WEBMD。自然药物综合数据库。网络。2012 年 10 月 28 日。<http://www.webmd.com/vitamins-supplements/ingredientmono-947-MARIJUANA.aspx?activeIngredientId=947&activeIngredientName=MARIJUANA>。
"10 种最常见的大麻使用健康副作用 了解更多信息:"http://www.testcountry.org/10-most-common-health-side-effects-of-using-marijuana.htm
苯海拉明,也称为异丙嗪,是一种抗组胺药物。它通常以结晶状白色粉末的形式存在,可溶于水和酒精。异丙嗪通常用于治疗过敏反应。
在体内,当人们遭受过敏反应甚至病毒感染时,组胺会释放。然后,组胺与细胞受体结合,触发细胞的改变,导致过敏症状,如瘙痒和打喷嚏。
作为一种抗组胺药物,异丙嗪与组胺竞争细胞受体。抗组胺药不仅阻止组胺与细胞结合并刺激细胞,而且在这样做的时候,它也与受体结合——它们不会刺激细胞,因此过敏症状不会出现。
苯海拉明是由乔治·里夫斯奇尔发现的,他在辛辛那提大学参与了许多专注于缓解肌肉疼痛的实验。苯海拉明是一种抗组胺药物,用于治疗患有过敏症的人。苯海拉明于 1946 年首次通过处方获得。后来在 1980 年代,它被批准为非处方药。
苯海拉明含有组胺阻滞剂异丙嗪。这种药物将帮助患者从常见症状中恢复:打喷嚏、流泪、流鼻涕和喉咙痛。苯海拉明的一种应用是苯海拉明胸闷咳嗽和鼻塞:一种用于缓解胸闷咳嗽和感冒症状的药物。
如果您目前正在治疗抑郁症、心脏病、血压和行为障碍,请勿使用这种药物。像许多其他药物一样,苯海拉明确实有一些常见的副作用。
- 嗜睡
- 眩晕
- 头痛
- 口干
- 排尿困难。
异丙嗪的合成如下所示。异丙嗪的分子式为 C17H21NO • HCl。 
"苯海拉明 (异丙嗪) 药物信息"。RXList。2009 年。检索于 2009 年 3 月 6 日
俄亥俄州历史中心:苯海拉明;访问时间:2011 年 1 月 5 日
适合家庭的苯海拉明胸闷咳嗽和鼻塞口服液;访问时间:2011 年 10 月 13 日
http://www.medicinenet.com/diphenhydramine/article.htm
维基百科共享资源中的图片

泼尼松是一种属于皮质类固醇类的药物,皮质类固醇是一种类固醇。类固醇是具有相似化学结构的激素,它们聚集在一起形成一个组。一般来说,泼尼松是由您的肾上腺产生的,肾上腺位于肾脏上方。它用于治疗皮质类固醇水平低的人。它通过阻止体内的易燃物质发挥作用。患有以下疾病的患者可以用泼尼松治疗:皮肤病、狼疮、关节炎、呼吸系统疾病或过敏性疾病,以常规剂量服用,或者可以用较高剂量治疗癌症。这种药物不推荐使用,因为它有非常严重的副作用。
Arthur Nobile 于 1950 年确定了泼尼松的结构。直到 1955 年,泼尼松的合成才在先灵公司实验室中完成;它是皮质酮的复制品——由肾上腺外层产生。如今,泼尼松可以作为非处方药开具。
泼尼松以片剂形式存在,可以直接口服。服用此药前应先吃点东西。您应按照规定剂量服用,因为过量服用会导致严重的副作用。您的医生将在您接受治疗期间调整您的剂量,但主要是您的身体可以接受的最低剂量。即使您感觉好转,也不要停止使用泼尼松,因为您的身体可能没有足够的来自这种药物的类固醇来正常运作。
泼尼松是一种糖皮质激素受体。它首先会被代谢为其活性形式,穿过细胞膜,并与特定的胞质受体结合。这会导致炎症的嗜中性粒细胞浸润、干扰炎症反应和抑制免疫反应。可以将泼尼松的抗炎和免疫抑制作用描述为以下几种情况
- 抑制基因转录
- 阻断骨钙蛋白
- 改变胶原酶基因的转录
- 增加膜联蛋白-1 的合成

您应该记住泼尼松是一种类固醇。它是一种非常有效的药物,如果使用得当,可以挽救生命。就像许多其他药物一样,它也会产生可能非常有害的副作用。使用这种药物时您可能会遇到的一些常见的轻微副作用是
- 头痛
- 头晕
- 睡眠障碍
- 皮肤过敏
- 极度疲倦
- 肌肉无力
其他人可能会遇到需要谨慎对待的严重副作用,并立即通知您的医生
- 视力问题
- 喉咙痛、发烧、咳嗽
- 皮疹
- 胃痛
- 呕吐
- 抑郁、意识混乱
- 呼吸系统疾病
http://www.gihealth.com/html/education/drugs/prednisone.html
http://www.drugs.com/prednisone.html
AHFS 消费者用药信息——泼尼松

烟草是一种作物,是香烟的主要成分之一。它被归类为兴奋剂。烟草是导致最严重依赖的药物。这种依赖源于其成分之一尼古丁。这种药物最常见的消费方式是吸烟。尼古丁是烟草中主要的活性成分。尽管它最常被用作娱乐性药物,但它已被证明具有医疗用途。烟草属于烟草属植物。这些植物中的尼古丁进化为抵御昆虫的一种机制。尝过这种植物的昆虫会受到神经毒素的攻击。在商业上,这些植物已被栽培和种植作为一种娱乐性药物,它对大脑起兴奋剂作用。

尼古丁的化学结构与神经递质乙酰胆碱非常相似。由于结构相似,尼古丁能够与受体结合并激活胆碱能受体,就像乙酰胆碱通常可以做的那样。当乙酰胆碱与这些受体结合时,它被认为可以刺激大脑功能并帮助控制肌肉。它在脑部处理速度中发挥着重要作用。尼古丁的结构相似性使其能够像乙酰胆碱一样激活这些受体。
咀嚼时,尼古丁很容易被肺部或口腔粘膜吸收。随着烟草的频繁使用,体内尼古丁水平在白天积累,并在夜间保持。因此,烟草使用者每天 24 小时暴露于尼古丁的影响之下。由于尼古丁的代谢和分泌取决于多种因素,即使在最后一次吸烟后的四天内,也可能在体内和尿液中检测到这种药物。
尼古丁具有以下行为影响
- 欣快感
- 精神兴奋剂
- 焦虑
- 肌肉放松
- 止痛药(减轻疼痛)
- 减少食欲。
如果在怀孕期间使用尼古丁,它也会产生不良影响。它有毒,会影响婴儿。如果女性吸烟,毒素和化学物质会进入她的血液,随后婴儿会通过血液获得这些物质。这是导致婴儿猝死综合征的原因之一。婴儿猝死综合征是指不满一岁的婴儿突然死亡,没有明显原因。尼古丁也可能导致流产和婴儿出生体重过低。因此,建议在怀孕期间不要吸烟,因为它会对胎儿产生不利影响。
Nesbitt 的悖论是关于尼古丁最著名的实验和观察之一。虽然尼古丁被归类为兴奋剂,并且在大多数情况下它像兴奋剂一样起作用,但尼古丁也被证明可以产生放松作用。尼古丁有时被描述为具有抗焦虑和抗压力特性。但与此同时,它会刺激大脑的功能。
烟草成瘾有三个阶段
| 阶段 | 效果 |
|---|---|
| 第一阶段 | 心理社会 |
| 第二阶段 | 愉悦 |
| 第三阶段 | 负强化“上瘾” |
烟草戒断的发生时间大约是:12-24 小时。它可以持续长达 10 天。烟草戒断的主要症状包括:强烈的渴望、易怒、焦虑、烦躁不安、睡眠障碍、食欲增加(主要是碳水化合物)、心率降低、新陈代谢降低和疲劳。当意识到烟草中的尼古丁会刺激人脑功能时,这些症状是有意义的。它会增加心率并帮助对抗疲劳。戒断随后会导致相反的不利影响。它会让你更渴望它,并让你更快地死亡。

由于有如此多的人使用烟草,治疗方法也多种多样。其中一种治疗成瘾的方法是认知行为疗法。这是一种心理治疗方法,可以让成瘾患者有机会谈论他们的成瘾。另一种人们使用的方法(虽然不那么频繁)被称为动机强化。这通常是指治疗师进行旨在激励患者戒烟的治疗课程。虽然这些治疗方法已被证明是有效的,但也有可以用来对抗成瘾的药物辅助。这里列出了一些例子
- 布propion - 它的商品名是 Zyban。这是一种典型的抗抑郁药。它的主要作用是去甲肾上腺素-多巴胺再摄取抑制。通过阻断它,它充当尼古丁拮抗剂。它不允许尼古丁与活性位点结合并启动与吸烟或烟草相关的正常行为改变。安慰剂试验表明,这种药物可以显着降低对尼古丁的渴望,并有助于阻止与烟草相关的戒断症状。这种药物被归类为具有低滥用潜力的药物。
- 伐瑞尼克 - 它的商品名是 Chantix。这是一种部分尼古丁拮抗剂。它以片剂的形式提供,可以每天一次或两次用水服用。这种药物基本上可以阻断尼古丁激活 a4b2 受体的能力,而 a4b2 受体负责尼古丁的作用。这种药物还可以帮助阻断尼古丁戒断症状。这种益处使其成为处理烟草成瘾时使用最多的药物之一。然而,最近的研究表明,一些人在服用这种药物时可能会表现出敌意、躁动、自杀念头和/或情绪低落。然而,这仅见于仍在吸烟的人服用伐瑞尼克时。重要的是要注意,这种药物不会立即起作用,可能需要长达 12 周才能完全消除成瘾。
除了药物辅助之外,尼古丁替代品也被证明有助于治疗成瘾
- 口香糖 - 这种口香糖有助于降低对尼古丁的渴望,并有助于阻止尼古丁戒断的不利影响。这是一种替代品,因为口香糖含有少量尼古丁。通过遵循预定的计划,咀嚼的尼古丁量越来越少,理想情况下会导致对香烟的渴望停止。然而,在极少数情况下,已经证明可能转变为对尼古丁口香糖的成瘾。
- 贴片 - 贴片是一种与口香糖类似的治疗方法。这些贴片通过皮肤释放受控量的尼古丁,这有助于阻止对尼古丁的渴望。理想情况下,它最终会导致对香烟的渴望停止。
- 无烟香烟 - 这些基本上是通过从香烟本身中去除焦油和其他有害化学物质而“清洁”的香烟。然而,烟雾进入肺部和肺癌的副作用并没有被证明会因无烟香烟而降低。无论如何,这是一种替代真香烟的方法。
- 阿尔茨海默病治疗 - 阿尔茨海默病是一种影响大脑的疾病。它会导致记忆、思维、认知和行为问题。烟草显示出改善认知能力的迹象。烟草中的一种成分“可替宁”已被研究并证明可以保护脑细胞。这非常重要,因为如果科学家能够保护脑细胞,它将减缓甚至预防阿尔茨海默病导致的脑损伤,并可能成为一种潜在的治疗方法。
- 止痛药 - 烟草已被证明可以提高疼痛阈值并减少疼痛。它可以潜在地用作医院的止痛药,以减轻患者的疼痛或在进入手术室之前提高他们的疼痛阈值。
- 体重控制 - 如前所述,烟草的行为影响之一是食欲下降。科学家正在努力研究并创造一种非成瘾性药物,通过使用烟草来降低对食物的渴望。
1. Hart, Carl. Drugs, Society, and Human Behavior. 13th. McGraw-Hill Humanities, 2008. Print.
2. Meeker-O'Connell, Ann. "How Nicotine Works" 2 January 2001. HowStuffWorks.com. <http://health.howstuffworks.com/wellness/drugs-alcohol/nicotine.htm> 2 December 2010.
3. Berg, Jeremy; Tymoczko, John; Stryer, Lubert. Biochemistry, 6th edition. W.H. Freeman and Company. 2007

酒精是一种精神活性药物/饮料。它是第二常用的精神活性药物。(咖啡因被评为最常用的精神活性药物。)它被认为是一种抑制剂,因为在中等或高浓度下,它会抑制神经元放电。(尽管在低浓度下,它可能产生相反的效果。)[6] 它不含任何维生素或矿物质。但是,它确实含有卡路里。需要注意的是,这种类型的酒精不应概括为与有机化学术语酒精相同。相反,这种类型的酒精,就有机化学而言,实际上是乙醇(或乙醇)。它的化学式为:CH3CH2OH。
由于其小而两亲的性质,乙醇可溶于水和脂肪,因此能够穿过细胞膜,使其能够轻易地对细胞造成损害并影响整个身体。[6]
在 10 世纪,一位名叫拉齐的波斯炼金术士发现了第一种酒精。今天,这种酒精被称为乙醇。最初,名称 kuhl 或 kohl 被赋予通过升华天然矿物辉锑矿制成的非常细的粉末,以形成称为硫化锑的化合物。如今,这种产品用作眼线笔和化妆品。
在 16 世纪,英文中出现了“酒精”一词,意思是“非常细的粉末”。威廉·约翰逊在他 1657 年的《化学词典》中将该词描述为 antimonium sive stibium。随着时间的推移,这个词逐渐演变成指通过蒸馏过程获得的任何液体。这种液体包括葡萄酒酒精。在 1594 年,利巴维乌斯在他的《炼金术》中将酒精称为 vini alcohol vel cinum alcalisatum。最终,这个词被发现意思是“葡萄酒烈酒”。直到 18 世纪,“葡萄酒烈酒”才被称为乙醇,并在今天的世界中扩展为“酒精”。
与其他药物不同,乙醇不需要消化。身体可以迅速将其吸收进入系统。大约 1/5 的酒精直接通过胃壁吸收。接下来,空胃的上部吸收它。酒精也可以通过肺部吸收。在 30 到 90 分钟内,身体能够从酒精饮料中达到最大血液浓度。
血酒精含量(或通常缩写为 BAC)是衡量一个人血液中酒精含量的指标。更具体地说,它是衡量一个人 100 毫升血液中酒精的克数。例如,如果一个人测得的 BAC 为 0.20,则该人每 100 毫升血液中含有 1/5 克酒精。
酒精主要通过代谢从体内消除。这主要发生在肝脏。一小部分(5-10%)通过尿液和呼吸直接消除。酒精的消除被认为是零级动力学。这意味着消除的酒精量随着时间的推移是恒定的,并且不取决于摄入的浓度。这与许多其他具有半衰期并且取决于体内含量的药物不同。酒精随着时间的推移不断消除。肝脏大约每小时可以消除 0.25 盎司。完全消除一杯(标准)酒大约需要两个小时。
酒精会增加伏隔核中多巴胺的释放。多巴胺负责运动、注意力、学习、情绪等。多巴胺也参与大脑的正性强化(奖励)系统。它还会触发内源性阿片类物质的释放。阿片类物质受体似乎也参与大脑的奖励系统。戒断症状非常强烈(例如,癫痫发作、NMDA 受体敏感性增强)。酒精对大脑奖励系统的神经影响是造成一个人对酒精生理性上瘾的主要原因。
从史前时代开始,一种被称为乙醇的化合物,通常存在于酒精饮料中,被人类食用已有多个世纪。摄入的原因是由于饮食、卫生、医疗、宗教和娱乐的原因(生命中发生的特殊事件)。醉酒的概念是指摄入大量的乙醇,导致醉酒。人们大量饮酒时经常看到的醉酒状态会导致宿醉,因为它的效果在一段时间后消退。重要的是要知道乙醇会导致严重的呼吸衰竭,甚至死亡,这取决于一个人摄入身体的多少。
乙醇的毒性是由主要代谢产物和次要代谢产物乙醛和乙酸引起的。据说所有伯醇都会分解成醛类。然后它被分解成羧酸,它们的毒性与乙酸和乙醛相似。服用如硫胺素等乙酸的受试大鼠的代谢物毒性降低。
此外,一些仲醇和叔醇不像众所周知的那样有毒。换句话说,这些醇类毒性比乙醇低,因为肝脏无法将其代谢成这些有毒的副产物。因此,它们更适合药用和娱乐目的。叔醇的一个很好的例子是乙氯维醇。乙氯维醇用于药用和娱乐目的。
然而,还有许多其他醇类的毒性比乙醇更强。原因是这些醇类需要更长的时间才能代谢,并且它们产生的物质比乙醇的毒性更大。一种木材酒精是甲醇。甲醇在肝脏中被氧化成甲醛,然后通过甲醛脱氢酶和醇脱氢酶被氧化成有毒的甲酸。糟糕的是,这会导致失明甚至死亡。在摄入乙二醇或甲醇后,服用乙醇是防止中毒的最佳方法。虽然甲醇被认为是有毒的,但它比乙醇的镇静作用弱得多。
通常,长链醇类,如异丙醇、正丁醇或正丙醇具有很强的镇静作用。它们的毒性也比乙醇高得多。这些长链也称为杂醇,令人惊讶的是,它们被发现会污染某些酒精饮料。因此,这是人们过度饮酒或大量饮酒会遇到宿醉的原因。总的来说,这些长链醇类甚至更长的链醇类在今天的工业中用作一种溶剂,并且被用于导致各种严重健康状况的酒精中。
在成瘾周期后,如果酒精摄入量减少或停止,可能会出现戒断。戒断症状通常在停止摄入后 24 到 48 小时出现。它会导致包括失眠、过度兴奋和震颤在内的症状。此外,它会导致交感神经系统发生变化,包括:心率加快、血压升高和体温升高。在这些早期的戒断症状出现后,晚期症状(通常在停止摄入后 2-4 天)可能会出现。这些包括震颤性谵妄发作、幻觉和高烧。除了身体变化之外,酒精戒断还会导致焦虑和烦躁症(消极情绪状态)。戒断的主要治疗方法之一是称为氯氮卓(利眠宁)的苯二氮卓类药物。它是一种镇静/催眠药物,有助于抵消酒精戒断的影响。
血酒精含量(或通常缩写为 BAC)是衡量一个人血液中酒精含量的指标。更具体地说,它是衡量一个人 100 毫升血液中酒精的克数。例如,如果一个人测得的 BAC 为 0.20,则该人每 100 毫升血液中含有 1/5 克酒精。
酒精主要通过代谢从体内消除。这主要发生在肝脏。一小部分(5-10%)通过尿液和呼吸直接消除。酒精的消除被认为是零级动力学。这意味着消除的酒精量随着时间的推移是恒定的,并且不取决于摄入的浓度。这与许多其他具有半衰期并且取决于体内含量的药物不同。酒精随着时间的推移不断消除。肝脏大约每小时可以消除 0.25 盎司。完全消除一杯(标准)酒大约需要两个小时。
酒精会导致抑制和判断力下降,从而导致攻击性行为。酒精还会导致感官障碍,并可能导致肝脏和心脏问题。
酒精会干扰神经粘附蛋白-蛋白,这些蛋白有助于引导发育中大脑的神经元生长。酒精是GABA受体的激动剂和NMDA受体的拮抗剂。GABA(γ-氨基丁酸)神经递质负责大脑的抑制,GABA受体激活氯离子通道。谷氨酸是NMDA受体的正常神经递质,但NMDA也可以以类似的方式激活这些受体。然而,NMDA不与其他谷氨酸受体结合。NMDA是一种兴奋性神经递质,负责学习和记忆。NMDA受体激活离子通道,允许钠离子和钙离子流入。
一些与饮酒相关的疾病包括柯萨科夫综合征和胎儿酒精综合征。柯萨科夫综合征的特点是维生素B1(硫胺素)缺乏,导致下丘脑和海马体的乳头体受损。
胎儿酒精综合征是指孕妇怀孕期间饮酒导致酒精损害婴儿发育。胎儿酒精综合征婴儿常见的两个特征是身体畸形和智力迟钝。它会导致轻度至中度智力迟钝。它还会导致儿童多动症以及生长缺陷。这种综合征还具有典型的面部畸形。乙醇对胎儿发育的影响包括但不限于:扰乱细胞粘附分子生成、扰乱正常凋亡(程序性细胞死亡)以及扰乱神经营养支持。[6]
酒精脱氢酶是将酒精转化为乙醛的主要酶。这种酶是酒精的主要代谢系统的一部分。酒精脱氢酶的活性决定了酒精代谢的速度。因此,运动或咖啡消费等说法在加快代谢系统方面没有作用。[5]
酒精脱氢酶缺乏会降低代谢过程的速度,因此分解酒精需要更长的时间。有证据表明,女性的酒精脱氢酶含量低于男性。有一些证据表明,女性月经周期、性腺激素水平的波动会影响酒精代谢系统的速度。女性在月经周期的不同时间可能会有较高的血酒精浓度。[7]


乙醛脱氢酶 (ALDH) 是脱氢酶,有助于将乙醛转化为乙酸。更准确地说,这种酶存在于代谢中,有助于分解酒精。当一个人饮酒时,酒精首先被酒精脱氢酶转化为乙醛。乙醛脱氢酶需要进一步将新的乙醛分解为更无害的乙酸。
如果一个人乙醛脱氢酶缺乏,这种分解无法发生,导致乙醛水平升高,从而导致呕吐、过度换气、恶心、心率加快和酒精冲红反应。研究发现,东亚人通常拥有编码乙醛脱氢酶2失活变体的基因。[2]
- “酒精:平衡风险和益处。”哈佛大学公共卫生学院(2010 年)。第 N 页。网络。2010 年 5 月 22 日。
- “酒精冲红信号表明东亚人患癌风险增加。”美国国立卫生研究院。2009 年 3 月 23 日。http://www.nih.gov/news/health/mar2009/niaaa-23.htm。
- “酒精饮料。” 维基百科,自由的百科全书。维基媒体基金会。2010 年 5 月 21 日。网络。2010 年 5 月 22 日。
- Carlson, Neil R. 行为生理学。波士顿:皮尔森教育,股份有限公司,2007 年。
- 哈特,卡尔。药物、社会和人类行为。第 13 版。麦格劳-希尔人文社科。2008 年。印刷。
- 皮内尔,约翰。“生物心理学”。皮尔森,第 7 版,2009 年。印刷。
- “酒精和女性”美国国立卫生研究院。1990 年 10 月。http://pubs.niaaa.nih.gov/publications/aa10.htm
- http://en.wikipedia.org/wiki/Alcohol#History_and_etymology
对乙酰氨基酚,又称扑热息痛,发音为“a set a mee” no fen”,是一种非处方 (OTC) 药物,由对氨基酚与乙酸酐反应制成。对乙酰氨基酚的通用名称是泰诺或MAPAP。对乙酰氨基酚是乙酰苯胺的衍生物。它用于暂时缓解轻度头痛、肌肉疼痛、喉咙痛和发烧。对乙酰氨基酚也可以用作治疗骨关节炎的抗炎药,骨关节炎是由关节错位导致的关节“磨损”引起的。这种药物通过改变身体感知疼痛的方式起作用,并产生反馈机制来冷却身体。这种药物不会阻断和抑制环氧合酶 (COX) 酶,环氧合酶是前列腺素产生的催化剂,而前列腺素反过来又负责在周围神经系统中产生疼痛、炎症和发烧,就像其他非阿片类止痛药一样。相反,人们认为对乙酰氨基酚会阻断中枢神经系统中的 COX-3。
虽然对乙酰氨基酚通常以片剂形式存在,但它也可以以溶液、悬浮液、咀嚼片、栓剂、滴剂等形式存在。虽然它是一种非处方药,可以轻松获得,但重要的是要知道服用它的风险和症状,就像服用任何其他药物一样。对乙酰氨基酚不应与酒精一起服用。过度使用对乙酰氨基酚会导致肝损伤,在严重但罕见的情况下,可能需要进行肝移植。此外,对乙酰氨基酚的报告副作用可能包括瘙痒、肿胀、声音嘶哑和呼吸困难,所有这些都应该考虑。如果症状持续,请停止服用药物并咨询医生。
将这种药物用于儿童可能非常有效且危及生命。用对乙酰氨基酚治疗儿童时,应极其小心地遵循说明和剂量。滴剂通常用于婴儿,但由于它们含有较高浓度的对乙酰氨基酚,请务必根据他们的体重和年龄检查并提供剂量。
此外,一些对乙酰氨基酚片剂含有额外的甜味剂,因此被诊断为苯丙酮尿症 (PKU) 的人应该注意和小心,因为他们的身体无法分解苯丙氨酸 (Phe),高水平的苯丙氨酸对诊断为 PKU 的人来说对人脑有害。
服用感冒药时,应避免服用像日夜百服宁和泰诺感冒等多种症状的感冒药,因为这些药物通常含有对乙酰氨基酚。相反,人们应该根据具体症状服用药物。例如,治疗鼻塞,建议使用速效感冒胶囊(通用名称为伪麻黄碱);止咳露用于咳嗽;泰诺用于发烧和疼痛;喉片用于喉咙痛等。
在实验室中,扑热息痛(对乙酰氨基酚)很容易通过硝化| 用硝酸钠硝化苯酚,从邻-副产物中分离出所需的 4-硝基苯酚| 对-硝基苯酚,并用硼氢化钠还原硝基来制备。然后用乙酸酐乙酰化所得的对氨基苯酚| 对-氨基苯酚。[1] 工业流程类似,但使用氢化代替硼氢化钠还原。[2]


另一种更简单的合成方法是 Hoechst-Celanese。这涉及用 HF 催化苯酚与乙酸酐直接酰化。从而将酮用羟胺转化为酮肟,然后进行酸催化的贝克曼重排生成酰胺。[3]
对乙酰氨基酚不太可能产生副作用。声称或出现对乙酰氨基酚引起的副作用症状的人,通常是由于对乙酰氨基酚过量引起的。
过量的常见副作用包括
- 恶心
- 黄疸(皮肤或眼睛发黄)
- 食欲不振
- 出汗
- 易怒
- 腹痛
- 肝功能衰竭
- 肾功能衰竭
- 心脏问题
- 昏迷
- 癫痫发作
- 死亡(在极端情况下)
http://pain.emedtv.com/acetaminophen/acetaminophen-side-effects.html
- ↑ Ellis, Frank (2002). Paracetamol: a curriculum resource. Cambridge: Royal Society of Chemistry. ISBN 0-85404-375-6.
- ↑ Anthony S. Travis (2007). “苯胺的生产和用途:各种各样的工艺和产品”。在 Zvi Rappoport (编辑). 苯胺化学 第 1 部分. Wiley. p. 764. ISBN 978-0-470-87171-3.
- ↑ Template:Ullmann
介绍
[edit | edit source]维柯丁,也称为扑热息痛和氢可酮,属于阿片类止痛药。阿片类药物有时被称为麻醉剂。其化学式为 C18H21NO3。扑热息痛和氢可酮的组合使扑热息痛因其效力较弱而增强了氢可酮的作用。在医疗用途上,维柯丁用于治疗剧烈疼痛。该片剂以维柯丁、维柯丁 ES、维柯丁 HP、Anexsia、Anolor DH5、Bancap HC、Zydone、Dolacet、Lorcet、Lortab 以及通用品牌等商品名生产和销售。研究人员表示,氢可酮比可待因强,但仅为吗啡与受体结合效力的十分之一。其镇痛作用约为吗啡的一半。但是,一些研究表明,氢可酮的效力在吗啡的一半到羟考酮(吗啡效力的 1.5 倍)之间,或者仅为羟考酮效力的 66.6% 左右。


上瘾
[edit | edit source]维柯丁成瘾是一种非常普遍的疾病,每年影响着许多美国人的生活,但只要接受适当的治疗,维柯丁成瘾是可以克服的。近年来,处方药滥用和成瘾现象呈上升趋势,但对成瘾问题的否认也持续存在。维柯丁成瘾是指对改变情绪药物的痴迷性滥用。在这个意义上,滥用意味着在没有医疗专业人士授权的情况下使用药物,或者在不需要的情况下继续使用处方药物。维柯丁成瘾会对一个人的身心产生压倒性的影响。
处方药成瘾,尤其是维柯丁成瘾会导致问题,因为它是第一次由医生开出的处方。在大多数情况下,那些对维柯丁上瘾的人会合理化并说“我是在服用止痛药”,或者“医生说我可以多服用几粒”。维柯丁会让人产生欣快感,放松身体和精神,并缓解疼痛。维柯丁成瘾在处方药使用者中极为普遍,最有可能的原因是它很容易被开具用于多种类型的疼痛问题。
一些处方药使用者并不打算对药物上瘾,但当他们的处方药用完时,他们开始痴迷于如何获得更多药物。维柯丁上瘾者发现,没有药物就无法正常生活,即使疼痛消失了,维柯丁也会对他们产生一种他们觉得不可或缺的作用。患有维柯丁成瘾的人开始寻找外部资源来获得更多处方,并且愿意不惜一切代价来获取更多药物。
维柯丁成瘾往往被认为比非法毒品或酒精成瘾不那么严重。这是因为维柯丁是由医生开的,而且维柯丁不像其他毒品那样会产生同样的负面问题。这种误解往往导致维柯丁上瘾者及其亲人相信他们不需要接受治疗。所有改变精神的物质都会影响大脑的运作方式,在维柯丁的情况下,大脑停止产生像内啡肽这样的化学物质。由于大脑没有产生这些必需的化学物质,身体就会感到无法在不使用药物的情况下正常运作。患有维柯丁成瘾的人已经对药物产生了依赖,如果他们无法获得更多药物,就会出现戒断症状和渴望。
从维柯丁成瘾中解脱出来是可能的。对于任何有毒品或酒精问题的人来说,寻求药物滥用治疗是最好的行动方案。在维柯丁成瘾治疗的初始阶段,上瘾者将经历排毒过程,以清除体内的药物,使他们为进一步治疗做好准备。我们的成瘾治疗机构将为上瘾者提供工具和资源,让他们在不使用维柯丁的情况下生活。
维柯丁是一种由扑热息痛和氢可酮混合而成的药物。氢可酮是一种作为麻醉性止痛药的药物,扑热息痛是一种比较温和的止痛药,可以帮助增强氢可酮的作用。
维柯丁可用于缓解中度至重度疼痛。服用维柯丁会损害判断力和思维能力,并降低警觉性。维柯丁不应与酒精一起服用,因为它会增加肝损伤的风险。
维柯丁应按处方服用,因为它具有成瘾性。一颗维柯丁片剂中含有高达 750 毫克的扑热息痛。扑热息痛过量会导致严重的肝损伤,甚至死亡。
服用药物过量的症状包括恶心、呕吐、胃痛、意识混乱、深色尿液、皮肤/眼睛发黄、心率减慢、呼吸浅表或肌肉无力。
服用维柯丁可能出现的一些严重副作用包括呼吸浅表、昏厥、癫痫发作、排尿困难、黄疸、情绪变化、视力模糊等。
与其他麻醉性止痛药、镇静剂、安眠药或其他可能引起嗜睡或呼吸减慢的药物同时服用维柯丁非常危险。 [1]
参考文献
[edit | edit source]http://www.drugs.com/vicodin.html http://www.rxlist.com/vicodin-drug.htm http://commons.wikimedia.org/wiki/File:Hydrocodone.svg http://commons.wikimedia.org/wiki/File:Hydrocodone_3d_balls.png “维柯丁”。WEBMD。2012 年 11 月 20 日。 http://www.webmd.com/drugs/drug-3459-Vicodin+Oral.aspx?drugid=3459&drugname=Vicodin+Oral&source=1
叶酸
[edit | edit source]
叶酸是 B 族维生素的一种,是产生红血球所必需的。叶酸是人工合成的叶酸形式;叶酸可以天然存在于某些食物中,例如绿叶蔬菜、坚果、豆类和谷物。一些谷物中含有女性每天应摄取的叶酸量的 100%。如果这种维生素含量不足,会导致贫血。由于人体自身产生的叶酸很少,服用维生素片形式的叶酸可以确保获得推荐的每日摄入量。
历史
[edit | edit source]在 1920 年代左右,许多科学家认为叶酸缺乏症和贫血是同一疾病。1931 年,一位名叫露西·威尔斯的研究人员确定叶酸是预防妊娠期间贫血所需的营养物质。换句话说,叶酸是怀孕期间必不可少的营养来源。通过这一鉴定,威尔斯博士表明贫血可以通过啤酒酵母逆转。因此,叶酸被认为是啤酒酵母中的矫正物质。1941 年,米切尔和其他人首次从菠菜叶中分离和提取了叶酸。
此外,1943 年,在美国氰胺公司勒德利实验室工作的鲍勃·斯托克斯塔德分离了纯结晶形式,随后确定了叶酸的化学结构。在研究主管兼负责人耶拉普拉加达·苏巴拉奥博士的指导和帮助下,一个被称为“叶酸男孩”的小组于 1945 年获得了纯结晶形式的叶酸。这个历史性的研究项目导致了抗叶酸氨蝶呤的合成。抗叶酸氨蝶呤是第一种抗癌药物。
然后,在 1950 年代到 1960 年代,许多科学家开始研究生化机制,并发现了叶酸的不同作用。这导致将叶酸缺乏症与神经管缺陷联系起来。总的来说,许多美国科学家注意到,市场上出售的食物中叶酸含量极少;因此,更多食物应含有叶酸,以帮助人们,尤其是孕妇。
含有叶酸的食物
[edit | edit source]许多健康食物富含叶酸。以下列出了一些。
- 蛋黄
- 葵花籽
- 肝脏和肾脏产品
- 绿叶蔬菜,如芜菁甘蓝、生菜、菠菜
- 豆类,如豆类、豌豆和扁豆
- 谷物产品,如意大利面、谷物、面包
含有少量叶酸的食物
- 水果,如香蕉、覆盆子、草莓
- 果汁,如橙汁或菠萝汁
请注意,叶酸天然存在于易受高温和紫外线照射的食物中。叶酸也是水溶性的。
叶酸与妊娠
[edit | edit source]叶酸已被证明可以预防怀孕初期出现的出生缺陷。这些出生缺陷包括脊柱裂,即脊柱和脊髓无法完全闭合的情况。它还可以预防无脑畸形,这是一种大脑无法发育的疾病。患有无脑畸形的婴儿通常在出生前或出生后不久就会死亡。 [1] 孕妇或正在尝试怀孕的女性建议每天服用 400 至 800 微克叶酸。
叶酸有可能将脊柱或大脑缺陷的几率降低近 70%。这些疾病被称为神经管缺陷,通常发生在脊髓在发育过程中未能完全闭合时,这就是脊柱裂。 [2]
叶酸最终会降低一种潜在有害化合物——高半胱氨酸的水平。它是通过加速高半胱氨酸转化为蛋氨酸来实现的,蛋氨酸是一种人体更喜欢和需要的无毒氨基酸。科学家和研究人员发现,将 MTHFR 酶锁定在其辅因子 FAD 上,可以让叶酸在人体内发挥其独特的功效。因此,美国食品药品监督管理局建议所有育龄妇女补充叶酸,以防止潜在的出生缺陷发生。
服用叶酸的副作用包括皮疹、瘙痒、发红或呼吸困难。
叶酸也可以通过均衡的饮食摄入。这包括强化谷物、全谷物、水果、蔬菜、豆类和其他天然蛋白质。那些因怀孕而想要增加叶酸摄入量的人可以通过饮食或补充剂和食物相结合的方式来实现。男性也要注意补充叶酸。虽然他们不能从预防出生缺陷中获益,但叶酸仍然是一种健康的补充剂,少量摄入是必要的。 [2]
精子质量
[edit | edit source]人们普遍认为,叶酸可以减少精子中的染色体缺陷。因此,叶酸是男性和女性生育的重要来源,因为它有助于精子生成。基本上,对男女而言,通过饮食获得足够的叶酸对于避免亚生育能力至关重要。
心脏病
[edit | edit source]使用叶酸会降低高半胱氨酸水平,但不会减少心血管疾病。但是,这对孕妇来说有所不同。怀孕期间摄入叶酸可以降低婴儿患心脏缺陷的风险,这是一件好事。它还可以降低儿童患代谢综合征的风险。
中风
[edit | edit source]尽管服用叶酸不会降低患心脏病的风险,但它似乎可以降低中风的风险。然而,许多评论表明,只有部分服用叶酸的人才能降低中风风险。换句话说,叶酸可能只对某些人有效。据称,中风减少与每天补充 5 毫克叶酸引起的脉压降低相一致。因此,对于可能患心脏病的人来说,在日常饮食中摄入叶酸至关重要。这也是为什么高半胱氨酸血症或中风患者的医生强烈建议他们每天服用包含叶酸的维生素 B。叶酸补充剂价格低廉,使用相当安全,但不要过量服用。
癌症
[edit | edit source]由于许多癌细胞耐受叶酸并过度表达叶酸受体,这导致了针对叶酸受体的抗癌药物的诞生。有研究证明,良好的叶酸水平可能与胃癌、食管癌和卵巢癌的风险降低有关。然而,叶酸对癌症的益处可能取决于个体服用叶酸的时间以及该人的具体状况。这是因为每个人都有不同的免疫系统,对某些事物的反应也不同。
此外,对于已经患有癌症或癌前病变的人来说,服用叶酸可能没有帮助,甚至可能是有害的。因此,摄入一定量的叶酸对日常饮食至关重要,但需要注意的是,过量的叶酸可能会促进肿瘤的发生。高叶酸摄入量会促进晚期致癌作用,而低叶酸摄入量会预防早期致癌作用。因此,许多医生和公共卫生机构建议在服用叶酸时要格外小心,并鼓励不要摄入过量的叶酸。
高叶酸饮食与结直肠癌风险降低有关。一些研究表明,从食物中获取的叶酸与从补充剂中获取的叶酸相比,其关联性更强。在叶酸和单碳代谢方面,结直肠癌是最常被研究的癌症类型。此外,流行病学研究表明,高叶酸饮食与乳腺癌风险降低有关。研究还表明,高膳食叶酸摄入量会降低患前列腺癌的风险。总体而言,已经进行过许多关于叶酸预防多种疾病的研究。
参考文献
[edit | edit source]- ↑ http://www.womenshealth.gov/publications/our-publications/fact-sheet/folic-acid.cfm
- ↑ a b 叶酸, 2012 年 10 月 28 日
http://en.wikipedia.org/wiki/Folic_acid [1] [2] [3]
定义
[edit | edit source]

阿亚华斯卡是一种植物致幻剂,南美洲亚马逊盆地的土著居民使用它。这种饮料在该地区的萨满教民族药学中占有核心地位。它是一种强大的神圣致幻剂,由阿亚华斯卡藤(Banisteriopsis caapi)和恰克鲁纳植物(Psychotria viridis)的叶子制成。Banisteriopsis caapi藤中含有 MAO 抑制剂,使Psychotria viridis中的精神活性物质 N,N-二甲基色胺 (DMT) 口服有效。这个名字本身就暗示了它的特性,阿亚华斯卡在克丘亚语中意为“灵魂之藤”,暗示了与宇宙精神本身交流的一种手段。
使用和一般效果
[edit | edit source]阿亚华斯卡主要用作一种药物或治疗工具,当地人认为它可以治愈任何疾病。它的制备方法是将Banisteriopsis caapi的树皮和茎与Psychotria viridis的叶子以及任何其他混合植物(Brugmansia spp.、Erythroxylum coca或Nicotiana rustica)一起浸泡和煮沸。当饮用这种饮料时,其主要效果是:一种清洁和治愈身心的大便(呕吐),伴随有幻觉以及一种改变的意识状态,据说可以让饮用者直接与阿亚华斯卡精神的更深层的精神智慧交流。饮用后大约 20 到 60 分钟开始起效,效果持续大约 4 到 8 个小时。传统上,这种饮料会由村庄里的治疗师或curandero饮用,他们会使用阿亚华斯卡来诊断和治疗那些来找他(或她)的人,无论是什么心理、精神或身体疾病困扰着他们。
Banisteriopsis caapi的主要生物碱是β-咔啉衍生物哈尔明、四氢哈尔明和哈尔马林。哈尔明和哈尔马林是高度可逆的单胺氧化酶(MAO)抑制剂,而四氢哈尔明则是突触前部位的弱5-羟色胺(5-羟色胺)摄取抑制剂。另一方面,Psychotria viridis含有单一的生物碱N,N-二甲基色胺(DMT),以及据报道为微量成分的N-甲基色胺和甲基-四氢-β-咔啉。β-咔啉的主要作用是抑制外周MAO,这可以保护阿亚华斯卡药剂中的DMT免受外周降解,使其口服有效,并使其穿过血脑屏障并激活大脑中的受体位点。此外,由于β-咔啉阻断了5-羟色胺的再摄取和MAO-A的代谢,体内5-羟色胺浓度升高。β-咔啉是MAO-A的高度选择性抑制剂,MAO-A是该酶的亚型,其首选底物是色胺类,例如5-羟色胺或DMT。当添加Brugmansia spp.或Nicotiana rustica等混合植物时,阿亚华斯卡中通常存在的精神活性生物碱也会因β-咔啉对MAO酶的抑制而增强。
被抑制的过程是MAO酶催化生物胺的氧化脱氨。氧化根据以下一般反应式进行:RCH2NH2 + O2 + H2O + MAO → RCHO + NH3 + H2O。这种酶广泛分布于脊椎动物和无脊椎动物的各种组织中,例如大脑、肝脏、小肠、心脏、肺、血浆和血小板。MAO系统作为一种解毒机制,保护神经系统和心血管系统免受生物胺的毒害,这些生物胺可能是有毒的,并且可能通过饮食摄入。这些胺通常是由芳香族氨基酸脱羧形成的。
阿亚华斯卡类似物是指代替阿亚华斯卡传统成分(Banisteriopsis caapi和Psychotria viridis)的植物或化学物质。类似物是指具有替代Banisteriopsis caapi中β-咔啉的MAO抑制行为的成分,以及替代Psychotria viridis中DMT的成分。
使用以下类似物可以找到与传统阿亚华斯卡产生类似效果的植物组合的示例
含有DMT的植物:Mimosa hostilis、Diplopterys cabrerana、Psychotria carthagenensis、Acacia maidenii、Anadenanthera peregrina
MAO抑制植物:Peganum harmala、Passiflora spp.
Callaway, J. C.,D. J. McKenna,C. S. Grob,G. S. Brito,L. P. Raymon,R.E. Poland,E. N. Andrade,E. O. Andrade,D. C. Mash(1998)Hoasca 生物碱对健康人体的药理作用。民族药理学杂志。待发表。
Grunwell, J. N. “南美阿亚华斯卡旅游。” 多学科迷幻药研究协会(MAPS)通讯 8.3(1998 年秋季):59-62。
Heaven, R.,Charing, H.G. "植物灵性萨满教。" 命运书籍。佛蒙特州罗切斯特。2006 年。82-87。
McKenna, D.,G. H. N. Towers & F. S. Abbott。(1984)南美洲致幻植物中的单胺氧化酶抑制剂:阿亚华斯卡的色胺和 β-咔啉成分。民族药理学杂志 10:195-223。
西地那非枸橼酸盐,俗称伟哥,是一种用于治疗勃起功能障碍和肺动脉高压(PAH)的药物。该药物的主要功能是抑制降解 cGMP 的酶,cGMP 是阴茎血流的调节剂。西地那非枸橼酸盐的医学用途包括性功能障碍、肺动脉高压和高原病。
1998 年,伟哥作为一种治疗男性勃起功能障碍(MED)的有效方法被推出。它的通用名为西地那非枸橼酸盐。它迅速成为世界上最受欢迎和最常被开出的药物之一。它的年销量每年超过十亿美元。西地那非是由辉瑞公司在英国的研究设施中工作的一组药物化学家合成的。它是在试图生产一种治疗冠心病的药物时偶然发现的。它成为 FDA 批准的首个口服治疗 MED 的药物。
西地那非枸橼酸盐在化学上被命名为 1-[[3-(6,7-二氢-1-甲基-7-氧代-3-丙基-1H 吡唑并[4,3-d]嘧啶-5-基)-4-乙氧基苯基]磺酰基]-4-甲基哌嗪枸橼酸盐。西地那非枸橼酸盐为白色至类白色结晶粉末,在水中的溶解度为 3.5 毫克/毫升,分子量为 666.7。伟哥(西地那非枸橼酸盐)被制成蓝色薄膜包衣的圆形菱形片剂
-
 伟哥的逐步合成
伟哥的逐步合成

伟哥(西地那非)在体内的机制包括保护男性阴茎海绵体中的环磷酸鸟苷(cGMP)免受 cGMP 特异性磷酸二酯酶 5 型(PDE5)的降解。
首先,阴茎海绵体中的氧化氮(NO)与鸟苷酸环化酶受体结合。这导致 cGMP 增加,从而促进海绵体动脉内膜垫的血管舒张。内膜垫的血管舒张允许更多血液流入阴茎的海绵体组织,从而导致勃起组织膨胀并产生勃起。西地那非是 cGMP 特异性磷酸二酯酶 5 型(PDE5)的强效和选择性抑制剂,PDE5 负责降解阴茎海绵体中的 cGMP。一旦 cGMP 水平下降,海绵体动脉内膜垫就无法扩张以允许更多血液流动。在没有性刺激的情况下,因此没有激活 NO/cGMP 系统,西地那非不应该引起勃起。
乙醇和西地那非在体内引起类似的反应。因此,医生不建议在服用该药时饮用酒精饮料。乙醇会稀释血液并扩张血管,这会导致头晕、心跳加快和血压降低。西地那非对身体有同样的影响,这会导致遭受这些影响的可能性大大增加。
它属于一类名为 PDE5 抑制剂的药物。西地那非的作用是抑制环磷酸鸟苷 (cGMP) 特异性磷酸二酯酶 5 型,这是一种负责降解阴茎海绵体中 cGMP 的酶。然后,阴茎海绵体中的氧化氮与鸟苷酸环化酶受体结合,导致 cGMP 水平升高,从而导致平滑肌松弛。西地那非的分子结构与 cGMP 相似,并作为阴茎海绵体中 PDE5 的竞争性结合剂,导致更多 cGMP 并更好地勃起。西地那非由肝脏酶代谢,并由肝脏和肾脏排泄。如果与高脂肪餐一起服用,吸收会降低;达到最大血浆浓度所需的时间会增加大约一小时,而最大浓度本身会降低近三分之一。
-
 PDE5
PDE5

该分子对温度和光线非常敏感,因此必须在 15-30 摄氏度的室温下保存,远离潮湿或高温。此外,如果长时间暴露在光线下,它会导致分子反应。
勃起功能障碍是一种性功能障碍,表现为在性交过程中无法维持阴茎勃起。由于阴茎勃起是血液涌入阴茎并保留在海绵状孔隙中的液压效应,勃起功能障碍是由循环系统问题引起的。ED 的原因之一是电压门控钾通道的改变,这些通道在去极化期间的动作电位中起着重要作用。但是,还有许多其他导致 ED 的原因,例如糖尿病、激素缺乏和神经系统问题。被认为可以治愈 ED 的主要药物是 Viagra、Cialis 和 Levitra,它们在制药市场上相互竞争。
一项研究表明,勃起功能障碍随着年龄的增长而增加。到 60 岁时约为 20%,尽管在 21 世纪,心理因素(如压力、抑郁和焦虑)、药物、疾病和伤害正在加速这一进程,比年龄早得多。据估计,美国 70% 的人口患有勃起功能障碍,这是由于肾脏疾病等潜在疾病引起的。而吸烟会使患病的几率增加 50%。令人震惊的是,只有 33% 的患者会寻求建议或医疗帮助,因为这是导致 35% 的关系破裂的原因。
在这种情况下,有效的治疗方法包括向阴茎注射药物,改变生活方式,减少香烟或酒精的摄入量。减轻体重,并遵循改善血液流动的运动计划也有帮助。真空泵也有帮助,因为它可以促进血液流动,使阴茎勃起。
如果您出现以下一项或多项症状,请立即停止服用 Viagra
- 荨麻疹
- 呼吸困难
- 面部、嘴唇、喉咙或舌头肿胀
- 视力丧失
- 耳鸣
- 胸痛
- 心律不齐
- 肢体肿胀
- 呼吸短促
- 视力改变
- 阴茎勃起超过 4 小时
- 头晕
不太严重的状况可能包括
- 鼻塞
- 头痛
- 记忆问题
- 胃部不适
- 背痛
http://www.drugs.com/sfx/viagra-side-effects.html
Delar, Anthony。 “与过量酒精一起服用 Viagra 可能对您的勃起产生负面影响。”ArticleBiz.com。2010 年。周六 2010 年 11 月 20 日。
http://www.articlebiz.com/article/532090-1-viagra-with-excessive-use-of-alcohol-may-affect-your-erections-negatively/
维基百科贡献者。"西地那非。" 维基百科,自由的百科全书。维基百科,自由的百科全书,2010 年 11 月 18 日。网络。2010 年 11 月 22 日。
http://en.wikipedia.org/wiki/Sildenafil
“伟哥。”Drugs.com。2010 年 10 月 18 日。周六 2010 年 11 月 20 日。
http://www.drugs.com/viagra.htm
Vollhardt, K. Peter C. “有机化学:结构与功能。”第五版。W.H. Freeman and Company, 纽约 2007 年。第 1176-1177 页

Vyvanse 是一种神经控制药物,用于治疗患有注意力缺陷多动障碍 (ADHD) 的患者,这些患者表现出记忆力丧失或难以集中注意力的症状。其结构由右旋苯丙胺和氨基酸 L-赖氨酸组成。ADHD 由大脑中一些天然物质引起,Vyvanse 可以帮助调节这些物质的含量,使其正常工作。Vyvanse 还帮助患者应对 ADHD 的特定症状;它可以增强注意力并减少冲动行为。
Vyvanse 由 New River Pharmaceuticals 开发,自 2008 年起上市。Vyvanse 可以直接口服,通常在早上服用,因为如果在下午晚些时候或晚上服用,它会导致睡眠困难。建议不要服用超过处方量的 Vyvanse,因为它会导致一些严重的副作用。医生可能会先给患者开低剂量,以观察其身体对药物的适应情况。在患者治疗期间,医生通常会调整剂量。除了服用 Vyvanse 作为完整治疗的一部分,患有 ADHD 的患者还应接受其他治疗。
由于 Vyvanse 会导致依赖或成瘾,因此应妥善使用。与其他药物一样,Vyvanse 也有一些常见的副作用
- 难以入睡
- 胃痛
- 流感、发烧、出汗
- 头痛、头晕、腹泻。
但对某些人来说,这种药物可能会产生一些严重的副作用
- 癫痫发作
- 视力问题
- 儿童生长缓慢
- 突发事件的恶化
如果出现这些副作用,应联系医生。
http://www.drugs.com/pro/vyvanse.html

结构生物化学使我们对 HIV 有了更深入的了解,并在抗击 HIV 药物的开发中发挥了关键作用;其中一种药物是齐多夫定或叠氮胸腺嘧啶 (AZT),它以 Retrovir 的名称出售,是首个获批用于治疗 HIV 的药物。AZT 是一种核苷类似物逆转录酶抑制剂,称为 NRTI。AZT 特别推荐用于患有 HIV/AIDS 的孕妇,因为在怀孕期间和分娩期间服用这种药物可以降低母婴传播的可能性。副作用包括头痛、恶心和指甲变色。更严重的副作用包括贫血和骨髓抑制。
一旦结构生物化学认识到 HIV 是一种逆转录病毒,以前在病毒中发现的蛋白质就成为了药物靶标。Retrovir 抑制了病毒中的一种酶——逆转录酶的作用,该酶可以将自身 RNA 复制成 DNA,使其快速复制并将自身编码到我们的基因组中(这就是为什么我们无法“治愈”艾滋病,直到我们找到一种方法来从我们自己的基因组中剔除病毒的 DNA)。如果逆转录酶无法生成双链病毒 DNA,那么它就无法将其 DNA 整合到我们细胞的遗传物质中。然而,HIV 会随着时间的推移而产生对 AZT 的耐药性,因此 AZT 与其他 NRTI 和蛋白酶抑制剂一起用于“药物鸡尾酒”中,以最有效地对抗 HIV 和艾滋病患者。
如果您出现以下一项或多项症状,请寻求医疗帮助
- 荨麻疹
- 嘴唇、面部、喉咙或舌头肿胀
- 呼吸困难
- 严重肌肉疼痛
- 苍白皮肤
- 容易瘀伤
- 不明原因的体重减轻
- 苍白皮肤
- 膀胱控制力下降
- 严重下背部疼痛
- 肝脏问题
- 胰腺炎
- 严重皮肤反应
不太严重的症状可能包括
- 失眠
- 轻度恶心
- 便秘
- 关节疼痛
- 头痛
如果与以下药物一起使用,Retrovir 可能会产生剧烈反应
- 阿霉素
- 更昔洛韦
- 干扰素α
- 苯妥英
- 利巴韦林
- 削弱免疫系统的药物
http://www.drugs.com/mtm/retrovir-injection.html 生物化学,第六版,Berg。 http://www.rxlist.com/retrovir-drug.htm http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000869

药物拮抗剂被设计为与受体结合,专门阻断或减弱药物的作用。拮抗剂本身没有任何生物学效应。通常,医生使用药物拮抗剂来帮助患者戒断或治疗过量服药的患者。通过服用拮抗剂,特定药物的受体被阻断,因此被削弱或完全关闭。其有效性取决于结合受体的性质。
配体可以与生化受体结合,进而被激活。在药物的情况下,药物充当配体。受体既可以是细胞内的,也可以是细胞外的。它们可以存在于细胞核和线粒体中,甚至存在于细胞膜上。配体可以通过与受体本身(活性位点)或变构受体结合来引起作用。变构位点是受体上不归类为活性位点的其他位点。它们都调节受体的活性。拮抗剂有助于阻断这些相互作用,并防止激动剂诱导的反应。当激动剂与受体结合时,它们会“开启”细胞反应。当拮抗剂与受体结合时,它们会“关闭”细胞反应。通常,拮抗剂的效率取决于药物被设计在受体上的结合位置。但是,在所有情况下,拮抗剂都被定义为本身没有生物学效应。由于这个因素,它在人类药物和戒断治疗中很有用。
竞争性 - 这些拮抗剂与目标配体或激动剂结合的相同位点结合。但是,它不激活受体位点,而是阻断它,因此目标配体或激动剂无法结合并激活该位点。
非竞争性 - 这些拮抗剂与变构位点结合,并从那里起作用。它不需要与目标配体或激动剂结合在同一个位点来阻断或减弱其作用。
非竞争性 - 非竞争性拮抗剂不能单独起作用。相反,它们需要配体或激动剂与受体位点结合,然后才能与变构位点结合并开始发挥作用。通常,这些拮抗剂对较高浓度的激动剂的阻断效果比对较低浓度的激动剂更好。
部分拮抗剂 - 部分拮抗剂可以与活性位点结合,但不能完全阻断受体的作用。但是,它确实降低了受体的最大潜力。
沉默激动剂 - 这些拮抗剂没有激活受体的能力。它们只是结合并阻断受体的激活。
逆向激动剂 - 它们与激动剂结合在相同的位点,但诱导与该激动剂相反的生物学反应。

纳洛酮 - 是一种阻断阿片类药物作用的药物拮抗剂。它通过注射给药。在许多情况下,纳洛酮用于对抗阿片类药物过量,例如海洛因过量。它由蒂巴因制成。它是一种μ-阿片受体拮抗剂,可以快速阻断戒断症状,这在过量服药的患者中尤其危险。纳洛酮还与其他药物一起使用以防止药物滥用。例如,纳洛酮存在于美沙酮中。美沙酮用于通过随着时间的推移逐渐减少阿片类药物的剂量来帮助患者戒毒。但它也具有成瘾性。因此,纳洛酮用于部分阻断美沙酮中的阿片类药物作用。
纳曲酮 - 它也是一种阿片类药物拮抗剂。此外,它可以有效地用作酒精依赖管理药物。然而,纳曲酮是口服给药的,被归类为竞争性拮抗剂。与纳洛酮相比,它的作用时间更长,不可逆转。因此,它更适合用作紧急解毒剂,而不是主要解毒剂。这种拮抗剂在μ-和κ-阿片受体上都有活性。最近的研究表明,低剂量的纳曲酮可能能够治疗克罗恩病。正在计划进一步的研究。
布prenorphine - 布prenorphine是一种部分阿片类药物拮抗剂。它与µ-和κ-阿片受体结合。布prenorphine是一种蒂巴因衍生物,在u和k受体点上是部分激动剂。它有助于阻断和减弱阿片类药物的作用。它与大脑中的受体紧密结合,使其在体内难以发挥阿片类药物的作用。
1. Hart, Carl. Drugs, Society, and Human Behavior. 13th. McGraw-Hill Humanities, 2008. Print.
2. http://en.wikipedia.org/wiki/Receptor_antagonist
3. Berg, Jeremy; Tymoczko, John; Stryer, Lubert. Biochemistry, 6th edition. W.H. Freeman and Company. 2007

酪氨酸激酶抑制剂是能够干扰酪氨酸磷酸化过程的药物,酪氨酸磷酸化过程是细胞表面附近细胞信号传导所必需的。
科学家们发现,许多疾病,癌症是其中一个主要疾病,利用这种途径作为疾病发展的关键步骤。癌症利用这种过程,就好像存在一个导致产生许多受体或产生受体及其生长因子的突变一样,信号变得更强,更一致。因此,如果科学家能够破坏这种信号转导过程,就有理由相信癌症的发展可以暂时停止或完全停止。这个概念导致科学家将这些进步称为“信号转导治疗”。
蛋白酪氨酸激酶(PTK)是专门负责细胞之间和细胞内信号传导的蛋白质。在这一类别下,还有受体蛋白酪氨酸激酶 (RPTK) 等蛋白质。这些蛋白质跨越细胞膜,有一个面向外的结构域(在细胞周围),还有一个在细胞内部的结构域。这种结构使其成为这种类型的蛋白质从细胞外部到细胞内部或反之进行信息传递的最佳选择。
1. 替尼泊苷 - 这些分子与底物竞争,与 ATP 非竞争。起初,科学家们发现了一些起作用的天然化合物,但它们的选择性和效力并不高。然而,经过一些小的化学调整,第一类替尼泊苷被创造出来。第一类替尼泊苷证明了有可能阻碍特定 PTK 的活性,而不会对细胞产生毒性或干扰正常的细胞过程。
2. STI-571 - 该药物最初被发现是一种阻碍酶 PTK Bcr-Abl 激酶活性的方法,该酶是慢性粒细胞白血病 (CML) 发展阶段的关键酶。STI-571 通过抑制剂与作为 PTK 的 ATP 结合区域一部分的氨基酸之间的相互作用起作用。该药物已经进行到临床试验,并显示出对慢性期白血病患者有效果。然而,患有晚期疾病的患者复发了。STI-571 也用于治疗其他疾病,例如胃肠道间质瘤。
3. BMS-354825 - 该药物用于治疗对 STI-571 治疗产生耐药的患者。该抑制剂对 Bcr-Abl 激酶蛋白和 Src 激酶蛋白都有效,并且比 STI-571 менее специфичен. 该药物与 STI-571 的区别在于它与蛋白质的活性状态结合,而 STI-571 通过与蛋白质的非活性状态结合起作用。
4. 吉非替尼 - 该药物与 ATP 位点结合,用于治疗非小细胞肺癌 (NSCLC)。该药物自 2002 年以来一直在临床中使用,但发现只对一小部分患者有效。这部分患者在激酶结构域中具有激活突变。吉非替尼本来应该是 EGFR 激酶结构域抑制剂。然而,发现肿瘤的存活并不完全依赖于 EGFR 激酶的使用。因此,有一段时间很难说吉非替尼是否无效是因为抑制了 EGFR,还是因为药物无法长时间占据受体。最近的研究表明,只有当受体在癌症的存活中起主要作用,或者当药物可以与其他信号转导剂结合以引起某些癌细胞自毁时,EGFR 激酶抑制剂才会有用。
5. AG 490 - 该药物用于抑制一种名为 Jak-2 的蛋白,这种蛋白在细胞因子信号传导中起着至关重要的作用。发现 Jak-2 蛋白的扩增行为与许多白血病、淋巴瘤和一些转移性癌症有关。Ag 490 抑制 Jak-2 途径,从而抑制致癌表型的表达。由于该药物不通过破坏信号传导或细胞生长来抑制 Jak-2 蛋白,因此它也与免疫疗法联合使用进行实验。尽管免疫疗法不直接与消除已存在的肿瘤有关,但它在患者摆脱大量肿瘤后,有助于维持无瘤状态。事实证明,AG 490 与白介素 (IL)-12 联合使用,可有效诱导免疫系统产生抗肿瘤反应。
两种抑制剂的作用机制都是通过结合激酶结构域的 ATP 结合位点来实现的。然而,由于结构上的考虑,它们的结合方式不同;STI-571 与比 PD173955 更多的氨基酸结合。因此,STI-571 只与激酶的特定非活性结构结合,而 PD173955 的结合则不太特异。PD173955 比 STI-571 更有效的其中一个原因是,PD173955 可以结合激酶蛋白的多种构象,并且对活化环的状态不太特异。另一方面,STI-571 需要激酶的特定非活性结构才能结合,因此结合起来不太容易。这使得科学家得出结论,即使某些 ATP 竞争性抑制剂具有相似的基本结构,但官能团的差异确实决定了该特定分子或药物将结合什么。
Levitzki, Alexander 和 Eyal Mishani。酪氨酸激酶抑制剂和其它酪氨酸激酶抑制剂。生物化学年度评论报告。网络。2011 年 10 月 29 日。<http://www.annualreviews.org/doi/abs/10.1146/annurev.biochem.75.103004.142657?url_ver=Z39.88-2003&rfr_dat=cr_pub%3Dpubmed&rfr_id=ori%3Arid%3Acrossref.org&journalCode=biochem>。抗菌素耐药性——也称为耐药性——是指细菌、病毒、真菌和寄生虫等微生物发生变化,使其对用于治疗其引起的感染的药物失效。当微生物对大多数抗菌素产生耐药性时,它们通常被称为“超级细菌”。这是一个主要问题,因为耐药性感染可能会致命,可以传播给他人,并给个人和社会带来巨大的经济负担。
由于目前人类和农业中大量使用抗生素,耐药性致病菌的数量有所增加。这种耐药性是自然选择的结果,其中对抗生素具有耐药性的病原体存活并继续繁殖,而没有耐药性的菌株被杀死。
1928 年发现青霉素时,人们认识到抗生素治疗多种传染病的有效性和便利性,并将其视为理所当然。现在,抗生素的使用已变得越来越普遍,因此导致了更耐药的细菌菌株的产生。
这些细菌已经变得对过去的药物更具耐药性,导致了新一代能够处理这些传染病的药物。人们意识到,这些药物中的许多药物将变得无用,因为细菌表现出的机制会导致它们进化成比过去更好的细菌。具有类似结构和基因的细菌能够通过积累编码耐药性的多个基因、在细菌的 3 个不同区域发挥作用的泵,以及这些细菌中已经存在的药物的相似性来进化,从而帮助它们产生耐药性。(Nikaido 2011)
某些细菌菌株对某些药物产生耐药性有很多原因。大多数耐药性起源于用于生产抗生素的细菌,因为它们需要对其自身产品具有耐药性,或者起源于存在于环境中的微生物,因此暴露于当今使用的过量抗生素中。
细菌基因可能发生突变,影响药物靶向的蛋白质。由于这种变化,蛋白质可能对药物的生化作用不太敏感。虽然这些突变在正常情况下可能不会遗传给下一代,但抗生素的存在会影响选择过程,只有利于具有这种突变蛋白形式的细菌。这种耐药性对人工合成的药物有效,这些药物不能通过酶被灭活。
某些药物更容易被某些细菌产生的酶灭活。编码这些酶的基因通常来自最初用于生产这些抗生素的细菌。酶促药物失活的例子可以在氨基糖苷的磷酸化/乙酰化/腺苷化以及 β-内酰胺酶的水解中看到。
氨基糖苷通过降低这些抗生素的净正电荷而失活。这些修饰酶,如 AAC(3)-11,将作用于属于酶的系统发育组的底物的第 3 位。由于在这些细菌中发现了抗生素生产微生物,因此它们已经在这些细菌的 DNA 中存在,这使得它们对这些氨基糖苷具有耐药性。(Nikaido 2011)
B-内酰胺耐药性是由质粒基因编码的 B-内酰胺酶引起的。这很成问题,因为它们对许多药物(如甲氧西林和类似的化合物)具有耐药性,这些药物能够通过不同的酶(如 Tem B-内酰胺酶和 AmpC)被水解。这些药物被重新设计成第二代和第三代,但 AmpC 酶也进化了,以抵消这些药物。不久之后,将不得不引入旧版本的药物来对抗像万古霉素这样的酶,万古霉素与细胞壁肽聚糖的前体结合,而不是抑制酶本身。(Nikaido 2011)
对耐青霉素肺炎链球菌的基因进行测序表明,靶蛋白正在作为嵌合蛋白(部分来自其他生物体)产生。由于抗生素靶标的变化,该药物无效。
R 质粒通常在细胞到细胞的转移中非常有效地转移,这是因为细菌细胞彼此密切相关,才使得它们之间能够进行细胞到细胞的转移。高度稳定的 R 质粒也使其在耐药性方面更有利。包含耐药性基因的 R 质粒能够将这些基因传递到任何一段 DNA,因为它们是由转座子组成的。这些 R 质粒包含一个独特的 59 个碱基的 3'-序列,称为整合子,它催化耐药性基因插入到一个兼容位点。通过此过程,更多耐药性基因能够传递给多种细菌,从而获得更高的耐药性。(Nikaido 2011) (Kaiser 2011)
宿主细胞经常大量丢失从克隆载体获得的 R 质粒。然而,大多数天然来源的 R 质粒是稳定的,当新的宿主细胞繁殖时,丢失的频率较低。这可以归因于这样一个事实,即天然质粒在其结构中含有“杀手”元件,当质粒丢失时,会杀死宿主细胞。(Nikaido 2011)
此外,基因可以通过突变、染色体外 DNA 转移(如接合,即通过菌毛转移 R 因子质粒)、通过从环境中获取 DNA 的转化以及通过噬菌体上的 R 因子的转导来获得耐药性。此外,基因可以通过使用转座子(或从一个 DNA 分子移动到另一个 DNA 分子的小型 DNA 片段)的转座耐药序列来对抗药物。[4]

如果药物与其靶点的接触受到限制,其有效性通常会降低。在局部,细菌可以产生某些蛋白质,这些蛋白质会影响核糖体或 DNA 的构象,从而限制抗生素进入这些靶标区域。这种机制的另一种方法是使用药物特异性外排泵。
减少药物进入细菌内部会诱导非特异性抑制。这种抑制是在细菌降低其外膜通透性,从而减少药物进入细菌的情况下产生的。通常,选择孔蛋白缺陷型突变体。作为一把双刃剑,外膜的较低通透性也会降低进入细菌的营养物质的摄入,从而对细胞有害。已经发现孔蛋白编码序列中的突变降低了大体积抗生素的渗透率,但对小的营养分子没有影响,使其能够以正常水平渗透。(Nikaido 2011)
下一种方法是通过多药外排泵,这些泵最初在 E. coli 和 P. areuginosa 中被发现。这些泵已在大多数临床革兰氏阴性细菌中被发现,它们与 P. areuginosa 具有相似的系统。多药外排泵由 3 个不同的部分组成,即耐药结节分裂外排蛋白、门控外膜和膜融合蛋白。(Nikaido 2011)(Aeschlimann 2011)
主要促进超家族是最大的转运蛋白家族,其中包含许多外排泵。在这个类别中,QacA 和 QacB 是第一个能够通过其结合位点容纳多种配体的 QacR 诱导复合物的泵。它们将能够结合通过膜进入的这些药物,然后被泵出系统。一些这些外排泵将始终泵送,其中需要一个阻遏物来阻止操纵子让泵继续其功能。(Nikaido 2011)(Aeschlimann 2011)
小型多药耐药家族具有不同的外排泵,这些泵在革兰氏阴性细菌的染色体上被编码并被观察到。这些泵的转运蛋白将遇到被去质子化并通过质子的内向通量泵出的底物。(Nikaido 2011)
下一类外排泵属于耐药-结节-分裂家族,其中这些泵与另外两类蛋白质相关联,即外膜通道蛋白和膜融合蛋白。这种泵的构建为细菌带来了巨大的优势,因为它可以直接将这些药物导出到培养基中,并从细菌外部,在那里它们必须通过外膜屏障重新进入细菌。Nikaido 2011)
这些泵将协同工作,产生协同作用,它们将泵出大多数抗生素并变得耐药。泵越多,耐药性就越强。虽然有些泵无法携带某些抗生素,但 AcrD 的同源物可以执行此功能,使细菌对抗菌剂的耐药性更强。(Nikaido 2011)
持久性细胞
[edit | edit source]当高浓度的抗生素被引入细菌时,可以安全地假设所有细菌都会被杀死。但当一些细菌细胞在抗生素中存活时,会发生一种特殊的现象。这些细胞被称为持久性细胞,它们已经成为细菌产生耐药种群的一种策略。细菌在种群中产生表型不同的混合物,以便其中任何一种都可以在改变环境因素时变得有利。因此,在存在这种持久性细胞的情况下,抗生素治疗被认为是无效的。

这些细胞类似于孢子,其中一小部分这些细胞在细菌种群中处于休眠状态。由于处于休眠状态,它们不会对抗生素产生反应,使它们能够在抗生素消失后重新感染。目前还没有针对持久性细胞的治疗方法,但有可能通过抗菌肽靶向这些细胞。这些 AMP 将靶向休眠或活跃的细胞,这将有效地攻击持久性细胞。(Nikaido 2011)(Duchene 2011)
参考文献
[edit | edit source]Nikaido, Hiroshi (2009). "细菌中的多药耐药性" (PDF). Annu Rev Biochemistry. Retrieved 2011-11-12.
Duchene, Ariel (2011). "解决细菌感染中持久性细胞的挑战". PHYSORG.com. Retrieved 2011-11-12.
Aeschlimann, Jeffrey R. (2003). "多药外排泵在铜绿假单胞菌和其他革兰氏阴性细菌抗生素耐药性中的作用". Medscape News. Retrieved 2011-11-12.
Kaiser, Gary E. (2001). "R质粒接合". Doc Kaiser's Microbiology. Retrieved 2011-111-7. {{cite web}}: Check date values in: |accessdate= (help)
- Nikaido H. (2009). 细菌中的多药耐药性。Annu. Rev. Biochem. 78, 119–146。doi: 10.1146/annurev.biochem.78.082907.145923.
- Spratt BG。靶点改变介导的抗生素耐药性。科学。1994;264:388–93。
Tortora, Gerard J., Berdell R. Funke 和 Christine L. Case。微生物学导论第 10 版。波士顿:本杰明·卡明斯 :, 2010。印刷。| 第 20 章 |
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000723/
- ↑ Berg, Jeremy M., ed. (2002), Biochemistry (6th ed.) 纽约市,纽约:W.H. Freeman 和 Company,
- ↑ PubMed Health,“脊髓脊膜膨出”。
- ↑ {Tortora, Gerard J., Berdell R. Funke 和 Christine L. Case。微生物学导论第 10 版。波士顿:本杰明·卡明斯 :, 2010。印刷。| 第 20 章| 第 600 页}
抗病毒药物的介绍
[edit | edit source]抗病毒药物是为了预防病毒感染而发明的。它们是抗菌剂中的一类,对人体无害。抗病毒药物不是破坏目标病原体,而是抑制在病毒感染中贡献一个或多个步骤的蛋白质。发明新的安全有效的抗病毒药物很困难,因为必须考虑对宿主细胞的危害以及病毒的变异。
四类抗病毒药物
- 神经氨酸酶抑制剂
- 蛋白酶抑制剂
- 中和抗体
- 基于蛋白质的融合抑制剂
神经氨酸酶抑制剂
[edit | edit source]神经氨酸酶的介绍
[edit | edit source]
神经氨酸酶,也称为唾液酸酶,是糖苷水解酶,可以裂解唾液酸的糖苷键。
神经氨酸酶有九种亚型。神经氨酸酶的主要类别有三种:
- 病毒神经氨酸酶
- 细菌神经氨酸酶
- 哺乳动物神经氨酸酶
唾液酸酶 1(溶酶体唾液酸酶),符号为 NEU1。
唾液酸酶 2(胞质唾液酸酶),符号为 NEU2。
唾液酸酶 3(膜唾液酸酶),符号为 NEU3。
唾液酸酶 4,符号为 NEU4。
为了更好地理解开发神经氨酸酶抑制剂的过程,将研究流感病毒神经氨酸酶抑制剂开发的案例。流感病毒的神经氨酸酶是一种锚定在病毒膜上的同四聚体糖蛋白。它分布在流感病毒的膜上,帮助成熟的流感病毒从宿主细胞中离开并感染新的细胞。对于每种流感病毒,在其表面大约有 100 个神经氨酸酶分布。

神经氨酸酶抑制剂的介绍
[edit | edit source]抑制神经氨酸酶会将新产生的病毒保留在宿主细胞中,并阻止感染其他细胞。
流感病毒分为两种类型,A型和B型。旧的药物,如金刚烷胺,只能有效地杀死A型流感病毒;对B型流感病毒无效。新的抗病毒药物通过改变药物上的官能团来减少耐药性,并改变它们与B型流感病毒的反应性,从而被发明出来。它们可以抑制A型和B型流感病毒。目前市面上有两种药物(扎那米韦,奥司他韦)。第三种药物仍在临床评估中。
扎那米韦是首个商业化开发的治疗流感病毒 A 和 B 的神经氨酸酶抑制剂。它由 Mark von Itzstein 领导的科学家于 1989 年发现。扎那米韦的分子式为 C12H20N4O7,分子量为 332.31 g/mol。扎那米韦没有已知的毒性作用。
奥司他韦是一种抗病毒药物,用于治疗和预防流感 A 病毒和流感 B 病毒感染。它于 1998 年 2 月获得批准。奥司他韦的分子式为 C16H28N2O4,分子量为 312.4 g/mol。它攻击流感病毒神经氨酸酶蛋白活性位点的疏水口袋,阻止病毒的复制过程。
蛋白酶抑制剂
[edit | edit source]
蛋白酶是一种存在于体内以裂解多肽的酶。特别是在 HIV 蛋白酶的情况下,这种酶是裂解 Gag 和 Pol 多肽所必需的,这对于 HIV 病毒的成熟至关重要。裂解的蛋白质被用来构建感染性 HIV 病毒体的组成部分。通过阻断这种蛋白酶,抑制剂阻止了 HIV 病毒的复制过程,导致 HIV 病毒体无法感染。设计一种抑制这种酶的药物很困难,因为蛋白质的动态特性控制着它与底物的结合能力。然而,由于 HIV 蛋白酶对 HIV 病毒的生命至关重要,因此它是设计新药的可行选择。
让我们先来看看 HIV-1 蛋白酶的结构,它是通过 X 射线晶体学发现的。

HIV 蛋白酶是一种对称的二聚体,具有一对活性位点残基 - 催化性天冬氨酸。此外,这些活性位点非常靠近对称轴。
有几种药物被用来控制 HIV 病毒和治疗艾滋病
安普那韦于 1999 年 4 月 15 日获得 FDA 批准。患者只需要每天服用两次药,而不是三次。安普那韦于 2004 年停止生产。

替拉那韦是一种由勃林格殷格翰生产的非肽类蛋白酶抑制剂,于 2005 年 6 月 22 日获得 FDA 批准。

达鲁那韦是第二代蛋白酶抑制剂,用于治疗 HIV 感染。达鲁那韦于 2006 年 6 月 23 日获得 FDA 批准。它的分子式为 C27H37N3O7S,分子量为 547.665 g/mol。
不幸的是,与许多其他药物一样,人类免疫缺陷病毒 (HIV) 蛋白酶抑制剂也遇到了耐药性和生物利用度方面的障碍。因此,讨论使用膦酸酯作为前药来解决这两个重要问题是相当合适的。吉利德集团一直致力于改善扎那米韦的生物利用度,他们发现了一种特定的膦酸二乙酯的共价结构,即 TMC-126,它是蛋白酶抑制剂安普那韦的类似物。膦酸二乙酯的一个重要特点是它能够穿透细胞膜。在穿过细胞膜后,膦酸二乙酯容易发生细胞内酶促水解,生成二膦酸二阴离子,后者不太可能从细胞中扩散出去。结果,这种化合物提高了 HIV 蛋白酶抑制剂在细胞内靶位点的有效浓度。另一方面,抑制剂的低耐药性可归因于 GS-8374 与非膦酸母体 TMC-126 结合热力学的变化。溶剂重排是该过程中的一个相当重要的过程,正如该过程的能量学所表明的那样,再加上膦酸与蛋白酶没有明显的相互作用。膦酸的结构也赋予了该分子在活性位点的空间灵活性,从而降低了突变的可能性。这种概念也可以应用于其他 HIV 蛋白酶抑制剂,而溶剂重排对于其他蛋白酶和治疗领域来说仍然是一个未定的概念。HIV 病毒膜抑制剂,如寡肽和恩夫韦肽,被证明在融合过程中作为螺旋束形成的破坏剂是有效的。然而,恩夫韦肽的成本和剂量方案使得对多药耐药的 HIV 患者进行治疗变得相当困难。药物设计人员现在正在寻找解决 HIV 患者对恩夫韦肽耐药性的方法。需要能够有效地竞争结合的相当大分子量的生物制剂来破坏相对较大的表面蛋白-蛋白相互作用,这限制了药物的其他方面。[1]

中和抗体
[edit | edit source]中和抗体 具有高度的特异性和高效性,可作为抗病毒药物。它们可以通过疫苗接种或先前的感染进入体内。由于抗体的特异性,研究表明,抗原的单氨基酸变化会导致抗体无法与抗原结合。在该研究后的结构研究表明,单氨基酸变化只导致局部结构变化。
虽然抗体在特异性地靶向病毒方面非常有效,但研究表明,病毒也具有机制来逃避抗体的检测。例如,在流感抗原中,酶促位点的呈现比抗体通常靶向的典型位点要小。HIV gp160 使用碳水化合物和构象变化来试图掩盖酶促位点。
目前,有一种单克隆抗体被注册为抗病毒药物:帕利维珠单抗,或 Synagis,在小鼠体内产生,用于儿科靶向呼吸道合胞病毒 (RSV)。在开发这种抗体时,选择了 RSV 融合体的三种特定变体进行体外测试:K272M、K272Q 和 N268I。
人们也对将这种疗法用于治疗丙型肝炎病毒感兴趣,但还需要进一步的研究。
基于蛋白质的融合抑制剂
[edit | edit source]这种类型的抑制剂基于阻止病毒膜与细胞膜融合,从而阻止病毒与细胞完全相互作用。通过抑制病毒的这种机制,细胞将不会被感染;因此,这是药物开发人员非常希望靶向的反应。这种反应的分子基础来自流感病毒的血凝素,即融合蛋白,在融合前和融合后的构象中。观察发现,膜融合过程遵循六螺旋束的产生。该途径中特异性的中间体是螺旋区域 1 (HR1) 中的三聚体螺旋卷曲,其中在 N 端发现的“融合肽”与靶膜相关联。
通过这种方法开发的首个药物是恩夫韦肽,它是一种寡肽,其序列与 HIV 融合蛋白 gp41 的螺旋区域 2 (HR2) 序列重叠。该药物的目标是通过竞争阻止螺旋束的形成,取代天然底物,从而创建一个屏障。然而,一些研究表明,它只弱地抑制六螺旋束的形成。药物的机制可能比预期的更复杂,需要进行更多研究。
新型抗病毒药物的设计
[edit | edit source]根据 Peter M. Colman 的文章“新型抗病毒药物和耐药性”,他宣布了一个假设,即药物与靶标的天然配体(底物)之间相似性的增加可以使病毒更难抵抗药物。除了这个因素,人们还应该考虑熵补偿和溶剂锚定来设计新的更好的抗病毒药物。
有几种方法可以改进药物,以对抗耐药病毒
- 提高药物浓度:如果药物能够广泛分布,并且也靠近受感染细胞中的靶标,那么药物将更有效。然后,浓度更高、分布更特异的药物可以使突变变得不那么重要。
- 向蛋白酶抑制剂添加带电荷的膦酸盐:科学家偶尔发现,含有膦酸盐的抑制剂可以与更广泛的耐药病毒变异体结合。GS-8373 就是一个例子,通过晶体学分析发现,膦酸基团在突变发生后仍然保持活性。没有膦酸盐的其他部分的活性降低了 10 到 40 倍。然而,根据 Peter M. Colman 的文章“新型抗病毒药物和耐药性”,科学家在含有膦酸盐的蛋白酶抑制剂的环境中培养病毒失败了。
- 提高药物与天然底物的相似性:形状、电荷和其他配置的相似性可以提高药物的有效性,使其与病毒结合以进行抑制。
参考
[edit | edit source]- ↑ Annu. Rev. Biochem. 2011. 80:239-46 The Annual Review of Biochemistry is online at biochem.annualreviews.org
1. Peter M. Colman, "新型抗病毒药物和耐药性"
2.邵华益,李卓荣,“神经氨酸酶抑制剂概述”
3.http://baike.baidu.com/view/555631.htm
4.http://baike.baidu.com/view/344762.htm
5.http://en.wikipedia.org/wiki/Neutralizing_antibody
6.http://commons.wikimedia.org/wiki/File:PDB_1hiv_EBI.jpg
介绍
[edit | edit source]氯胺酮是一种分离性麻醉剂,于 1963 年开发,用于替代苯环利定,目前用于人类麻醉和兽医。在街头出售的大多数氯胺酮都从兽医办公室中提取而来。氯胺酮的化学结构和作用机制与苯环利定相似。虽然它被制造为可注射液体,但在非法使用中,氯胺酮通常被蒸发形成粉末。使用方式是鼻吸或吞服。氯胺酮无色无味,因此可以混入饮料中而不被察觉,并且会引起健忘。由于这些特性,这种药物有时会被人给不知情的受害者服用,并用于性侵犯,称为“迷奸药”。
氯胺酮的短期影响是它可以引发梦境状态和幻觉。使用者报告的感觉从愉悦的漂浮感到与身体分离。一些氯胺酮体验包括一种可怕的几乎完全感觉分离的感觉,类似于濒死体验。这些体验类似于 LSD 的“糟糕旅行”,被称为“K 洞”。低剂量中毒会导致注意力、学习能力和记忆力下降。在较高剂量下,氯胺酮会导致谵妄、健忘、运动功能障碍、高血压、抑郁和可能致命的呼吸问题。
概述
[edit | edit source]氯胺酮是一种分离性麻醉剂。它被设计为一种弱酸性(pH 值 3.5-5.5)溶液,通过注射给药。它的化学名称是 (±)-2-(邻氯苯基)-2-(甲氨基)环己酮盐酸盐。氯胺酮在街头有各种名称。它被称为 K、Special K、猫镇静剂或维生素 K。氯胺酮被滥用,无论是液体形式还是固体形式。液体形式通常静脉注射,这是最危险的服用方式。固体形式用于吸食、鼻吸或吞服作为药丸。此外,已知它被添加到饮料中。氯胺酮被合法用作动物和人类的麻醉剂,但它通常被滥用来寻求快感。 [1]
效果
[edit | edit source]氯胺酮有许多强大的效果,使其成为一种受欢迎的私人用药。它的一些心理效应包括脱离身体的体验、幻觉、麻木、欣快感和恶心。所有这些效果都会随着剂量和耐受性的不同而不同。氯胺酮的影响可以持续长达 24 小时,但幻觉仅持续约 45 到 90 分钟。氯胺酮使用与严重的精神和身体损害有关。这些影响可能包括:谵妄、抑郁、高血压和运动功能丧失。氯胺酮还被用作迷奸药,使使用者失去意识。它有时会被掺入其他药物中,或被不了解药物强度的使用者服用。 [2]
药理学
[edit | edit source]氯胺酮是一种非竞争性 NMDA 受体拮抗剂。据信,阻断该受体会导致服用氯胺酮时出现的镇痛作用。
参考文献
[edit | edit source]http://www.abovetheinfluence.com/facts/drugsketamine http://dancesafe.org/drug-information/ketamine http://www.thegooddrugsguide.com/ketamine/basics.htm
MDMA
[edit | edit source]MDMA(3,4-亚甲二氧基-N-甲基苯丙胺)通常被称为“摇头丸”、“E”、“X”或“XTC”。它是一种用于产生“感觉良好”情绪的药物。它是一种具有兴奋作用的致幻剂。具体来说,MDMA 的结构与甲基苯丙胺(它获得“兴奋剂”特性的药物)在化学上相似[5]。兴奋剂是一种能够通过增加敏感性引起身心变化的精神活性物质。例如,使用者可能会对看似正常的事情感到更加快乐或愉悦,而那些没有受到影响的人则不会。他们的身体会对触摸和质地更加敏感,同时感到亢奋或精力充沛。MDMA 在化学上也类似于麦角碱,麦角碱属于致幻剂中的迷幻分支[5]。迷幻药往往会以影响思维、感知和意识的方式改变思维。MDMA 是一种非常受欢迎的药物,现在仍在使用。
历史
[edit | edit source]1912 年,默克制药公司合成了 MDMA,最初用作食欲抑制剂。随着时间的推移,在 1970 年代,MDMA 被用来增强沟通能力,据说它还被美国政府测试过,作为一种可能的“吐真剂”[6]。1987 年,MDMA 成为一种非法药物,因为人们怀疑它会导致使用者的大脑损伤,而这在使用这种药物进行的研究实验中使用的动物模型中是事实 [6]。虽然它是一种非法药物,但世界各地许多人一直在使用 MDMA。它通常在狂欢派对、俱乐部或聚会上使用。随着时间的推移,它已成为一种非常主流的药物。
效果
[edit | edit source]主观效应
[edit | edit source]MDMA 主要影响大脑中的神经递质,特别是血清素、多巴胺和去甲肾上腺素。MDMA 会增加血清素的释放,血清素是一种调节情绪、睡眠、疼痛、情感、食欲和其他行为的神经递质。据说 MDMA 会增加血清素的活性,并减少多巴胺的释放。血清素增加的原因是 MDMA 会抑制血清素进入再摄取部位,导致血清素过度流动。MDMA 对人的心理有许多影响。一些副作用包括欣快感、攻击性降低、失去自我意识、感官增强和对他人同理心的增强。MDMA 还有许多身体副作用。由于 MDMA 会增加血清素的活性,大脑会耗尽血清素。MDMA 使用者可能会经历许多使用该药物的副作用。一些身体副作用包括恶心、过度出汗、磨牙、心率加快、瞳孔散大、血压升高和体温升高。
后遗症
[edit | edit source]在使用药物后,使用者“从兴奋状态中恢复过来”时,他们可能会感到以下急性的后遗症:
- 心理方面
- 疲倦 [10]
- 抑郁 [10]
- 焦虑 [8]
- 注意力、专注力和集中力下降 [11]
- 同理心和亲密感的残留感觉 [8]
- 身体方面
- 头晕或头昏眼花 [12]
- 使用后几天内情绪波动 [12]
- 疲倦 [12]
- 抑郁 [12]
用法
[edit | edit source]
娱乐性使用
[edit | edit source]MDMA 广为人知的是在电子舞曲音乐 (EDM) 活动,更具体地说是在被称为“狂欢派对”的场景中用于娱乐。然而,许多用户喜欢在其他音乐活动中服用摇头丸,例如音乐会、舞蹈和派对场景,因为服用摇头丸似乎可以增强对音乐的感知。MDMA 通常以药丸的形式服用,但也可以被研磨并吸食。药丸的纯度通常是未知的。有许多种药丸,它们含有不同浓度的 MDMA,有时还会掺入其他药物,例如咖啡因和甲基苯丙胺 [7],从而改变用户体验到的药物效应。服用后,药丸通常在 30-40 分钟内起效,这通常取决于用户的代谢模式,而“峰值”效应通常在服用后约 60-90 分钟出现。用户通常会体验到 MDMA 的效果总共 3-4 小时 [12],不包括后遗症。
尽管 MDMA 因其娱乐用途而臭名昭著,但很少有人知道 MDMA 作为一种药物的前景。强调其精神活性作用及其提升情绪和精神状态的能力,MDMA 已在患有轻度精神疾病的患者、患有创伤后应激障碍 (PTSD) 的患者、癌症患者,甚至需要帮助与伴侣沟通的患者身上进行过测试 [8]。此外,Michael C. Mithoefer、Mark T. Wagner、Ann T. Mithoefer、Lisa Jerome 和 Rick Doblin 对患有 PTSD 的患者进行的 MDMA 研究得出结论:“MDMA 辅助心理治疗可以用于治疗创伤后应激障碍患者,并且没有证据表明其有害,并且可能对其他治疗无效的患者有用”[9]。
在 MDMA 被政府禁用之前,它在精神病医生中很流行,用于治疗医学研究,特别是在打开心灵以进行调查方面。在陆军的化学中心,MDMA 被用来减轻患有创伤后应激障碍的退伍军人的恐惧。据推测,MDMA “减少了对情感威胁的恐惧反应......许多受访者报告了 MDMA 的治疗益处。他们用它来揭开被压抑的痛苦的童年记忆和经历;减少恐惧和防御;增强与配偶的沟通和同理心;度过强奸和乱伦等创伤经历;忍受癌症的痛苦;坦然接受死亡。”这种新发现的安全感旨在让精神病医生能够轻松地与患者交流,而无需情感上的紧张。根据 Rosenbaum 和 Doblin 在《犯罪、法律与正义研究》第 7 卷中的研究,“创伤受害者接受了 MDMA 辅助心理治疗,帮助他们深入了解问题的根源,体验治疗性的宣泄,并随后更有效地运作。”然而,由于 DEA 将 MDMA 列入“附表 I”,因为 MDMA 有可能被滥用,尽管它被用作娱乐药物的倾向不受影响,所以 MDMA 的这种积极的治疗作用消失了 [14]。也就是说,许多研究人员由于 MDMA 被列入附表 I 后该药物所带来的负面含义,失去了继续研究 MDMA 治疗作用的兴趣,尽管在 MDMA 被禁期间,有 200 名医生使用 MDMA 进行心理治疗 [15]。此外,由于 MDMA 被宣布为非法,获得 MDMA 的难度更大,价格也更贵,这也是 MDMA 研究放缓的两个原因 [13]。
正式 IUPAC 名称:N-甲基-1-(3,4-亚甲二氧基苯基)丙烷-2-胺
其他名称:3,4-亚甲二氧基甲基苯丙胺、亚甲二氧基-甲基苯丙胺或 N-甲基-1-(1,3-苯并二氧杂环戊烯-5-基)-2-丙胺
MDMA 可以以 R 型和 S 型的外消旋混合物的形式存在。
MDMA 属于苯乙胺类,也称为“设计药物”,与苯丙胺类密切相关。苯乙胺类以其致幻作用而闻名,其致幻作用源于该化合物能够释放大脑中的血清素,以及能够刺激中枢神经系统,使使用者“更有”能量。MDMA 还有许多同源形式,包括:MDA、MDEA 和 MBDB。MDMA 主要以盐的形式存在;盐酸盐或磷酸盐,以片剂形式存在。它也可以以粉末或胶囊形式存在 [16]。作为碱性 MDMA 是一种无色油。
MDMA 由黄樟油生产,黄樟油是从黄樟树中提取的一种液体。将黄樟油转化为 MDMA 最常用的方法是通过瓦克尔法合成 3,4-亚甲二氧基苯基-2-丙酮 (MDP2P) 中间体,或者通过强碱将黄樟油异构化为异黄樟油。这两种方法都将黄樟油或异黄樟油氧化成 MDP2P 中间体,然后进行还原胺化,生成 MDMA 的外消旋混合物。
MDMA 由黄樟油生产,黄樟油是从黄樟树中提取的一种液体。将黄樟油转化为 MDMA 最常用的方法是通过瓦克尔法合成 3,4-亚甲二氧基苯基-2-丙酮 (MDP2P 或 PMK) 中间体(即乙烯在水中用四氯钯酸盐催化剂在氧气作用下氧化成乙醛 [17]),或者通过异构化(改变化合物中原子排列)将黄樟油转化为异黄樟油通过强碱。这两种方法都将黄樟油或异黄樟油氧化成 MDP2P 中间体,然后进行还原胺化,生成 MDMA 的外消旋混合物。在默克专利中,将黄樟油加入到 HBr 中形成溴黄樟油,然后加入甲胺,生成外消旋 MDMA [16]。其他合成方法从 3,4-亚甲二氧基苯基-2-丙酮开始,并使用 Leuckart 法、铝箔法或其他还原胺化方法 [16]。Leuckart 法使用甲酸、甲酸铵或甲酰胺/甲酰甲胺将酮转化为胺基。铝箔法使用乙醇、铝金属片、胺和氯化汞催化剂与酮反应。铝箔法是一种还原胺化(还原烷基化)方法,即使用亚胺中间体将羰基转化为胺基。
MDMA 与人体相互作用的主要方式是通过大脑。如前所述,MDMA 会诱导血清素水平升高,但也会释放多巴胺和去甲肾上腺素。MDMA 的结构使其成为血清素的间接激动剂,诱导神经递质的产生并抑制再摄取 [19]。事实上,MDMA 似乎作为所有这些神经递质的再摄取抑制剂发挥作用。MDMA 通过两种主要途径代谢:“1. O-脱甲基化,随后是儿茶酚-O-甲基转移酶 (COMT) 催化的甲基化和/或葡萄糖醛酸/硫酸结合;以及 2. N-脱烷基化、脱氨基和氧化为相应的苯甲酸衍生物,与甘氨酸结合”。CYP2D6 酶的反应性是 O-脱甲基化途径中的一个重要步骤,因为它调节 MDMA 的降解,并且该酶的效率(由基因编码)可能会增加使用者患急性毒性的风险。此外,MDMA 的代谢也可能与中长期神经毒性有关,因为神经传递系统会发生神经退化 [18]。
通常,药代动力学是药理学的一个分支,它处理药物与身体相互作用的不同阶段的时间安排。生物利用度一词定义为与药物的生物化学相互作用,用于预测身体将吸收多少药物。MDMA 的生物利用度可以通过血浆中该物质的浓度、尿液中的浓度甚至血浆中皮质醇激素的浓度来衡量,因为 MDMA 会诱导其出现。M. Farré 等人(2004 年)进行的研究绘制了 MDMA 非常复杂的过程图,以及在两次剂量间隔 24 小时后身体实际吸收了多少药物。在相应的出版物中:“人类重复剂量服用 MDMA:药理作用和药代动力学”表明,实验结果表明 MDMA 的浓度和药理作用显着增加,这表明第二次剂量对代谢具有抑制作用。MDMA 的“积累”也可以通过观察其结构中存在的亚甲二氧基来解释,据推测,该基团可以对 MDMA 的代谢产生“自抑制”[19]。据推测,MDMA 通过 CYP2D6 酶抑制其代谢。CYP2D6 酶是代谢进入人体的许多物质(包括维生素和药物)的一个重要因素。CYP2D6 酶通过肝脏中的氧化作用代谢物质。然而,对结构与 MDMA 相似的物质的研究表明,MDMA 可能能够通过亚甲二氧基形成酶-代谢物复合物,从而抑制 CYP2D6 酶的催化作用 [19]。MDMA 的这些“积累”和抑制效应证明,它表现出非线性药代动力学,由于身体无法以有效的速度清除该物质,因此可能产生极度有害的药理作用和毒性 [20]。
药理遗传学是药理学的一个分支,它关注代谢物质的酶和其他机制背后的遗传学。该重点领域对于确定 MDMA 对使用者的影响至关重要。目前正在研究负责 MDMA 代谢的 CYP2D6 酶的遗传学,以确定人体代谢 MDMA 的效率。分析人体吸收和处理 MDMA 的能力非常重要,因为它可以回答关于对死亡的易感性、药理作用的强度、毒性的强度和对依赖的易感性的问题。由于最近的研究表明 MDMA 具有非线性药代动力学,因此体内代谢 MDMA 的速率对于理解毒性、死亡率和药理作用至关重要。
遗传学影响物质代谢的一个例子是 Y. Ramamoorthy 等人 (2001) 的一项有趣研究,该研究表明,亚洲人 CYP2D6 底物的代谢率低于白种人,因为负责 CYP2D6 合成的基因存在变异,与典型的野生型基因不同,野生型基因编码的酶功能更慢 [21]。再次考虑到 MDMA 可能作为积累剂和抑制剂,那么如果亚洲人观察到的 CYP2D6 酶功能更慢,那么具有这种基因型的使用者患急性中毒和死亡的风险更高。然而,对于具有野生型基因型的白种人,该基因型产生具有正常快速功能的 CYP2D6 酶,那么他们对死亡的易感性和药理作用的强度低于亚洲人。在查看数据时,重要的是要记住,遗传群体仍然是人口的平均值,每个个体都有不同的遗传构成,这会影响药物的药代动力学。
1. http://upload.wikimedia.org/wikipedia/commons/b/ba/MDMA.png
2. http://www.nevamo.com/ecstacy.htm
3. http://www.drugaware.com.au/Drug%20Information/Ecstasy/Origin%20and%20How%20It%20Is%20Used.aspx
4. http://alcoholism.about.com/cs/ecstasy/f/mdma_faq05.htm
5. http://www.drugabuse.gov/publications/drugfacts/mdma-ecstasy
6. http://inventors.about.com/library/weekly/aa980311.htm
7. http://www.ecstasydata.org/
8. http://en.wikipedia.org/wiki/MDMA
9. http://www.maps.org/w3pb/new/2010/2010_Mithoefer_23124_1.pdf
10. http://www.urban75.com/Drugs/e_guide.html
11. http://www.communitybuilders.nsw.gov.au/download/ecstasy.pdf
12. http://www.erowid.org/chemicals/mdma/mdma_basics.shtml
13. http://www.psychedelic-library.org/rosenbaum.htm
14. 犯罪、法律与司法研究,第 7 卷,作者罗森鲍姆和多布林
15. 格伦·R·汉森,彼得·J·文图雷利,安妮特·E·弗莱肯斯坦(2005 年 11 月 3 日)。“毒品与社会(第九版)”。琼斯和巴特利特出版社。 ISBN 978-0-7637-3732-0。于 2011 年 4 月 19 日检索。
16. http://www.emcdda.europa.eu/publications/drug-profiles/mdma
17. http://en.wikipedia.org/wiki/Wacker_process#cite_note-1
18. de la Torre R, Farré M, Roset PN, Pizarro N, Abanades S, Segura M, Segura J, Camí J. “人类 MDMA 药理学:药代动力学、代谢和处置”。NCIB
19. R. De La Torre, M. Farré, J. Ortuño, M. Mas, R. Brenneisen, P. N. Roset 等人。(2000 年 2 月)。“MDMA(‘摇头丸’)在人体的重复剂量给药:药理作用和药代动力学”。施普林格出版社,2004 年。
20. R. De La Torre, M. Farré, J. Ortuño, M. Mas, R. Brenneisen, P. N. Roset 等人。(2000 年 2 月)。“MDMA(‘摇头丸’)在人体中的非线性药代动力学”。《纽约科学院院刊》,49(2):104-109。
21. Ramamoorthy,Y., Tyndale,R.F., and Sellers, E.M.(2001).Cytochrome P450 2D6.1andcytochrome P450 2D6.10differincatalyticactivityformultiplesubstrates.Pharmacogenetics 11, 477–487.

可卡因(苯甲酰甲基芽子碱)是一种结晶的托品碱,是中枢神经系统兴奋剂、食欲抑制剂和局部麻醉剂。可卡因源自古柯植物,最纯净的形式是珍珠白粉。该药物过去曾用作麻醉剂,但现在已被其他合成麻醉剂(如苯佐卡因)所取代。如今,可卡因主要用作娱乐性药物,由于它影响大脑的奖赏途径,因此具有高度成瘾性。
可卡因可以溶解在水中注射,以盐酸盐的形式吸入,以岩石晶体的形式吸烟,或以栓剂的形式阴道或肛门注射。吸入可卡因时,药物会穿过鼻子,并通过鼻腔组织吸收到血液中。为了注射可卡因,使用者首先将药物与水混合,然后用针头注射到血液中。这种方法存在艾滋病毒/艾滋病的风险,因为可能存在共用针头。吸食可卡因涉及将药物加工成岩石晶体形式。然后加热岩石晶体,使蒸汽被吸入。吸入后,可卡因进入肺部,并吸收到血液中。肛门和阴道注射可卡因使用口服注射器。将药物溶解在水中,插入注射器,然后通过肛门或阴道壁的膜内衬吸收。
说到纯度,可卡因通常是白色和珍珠色的。像盐酸可卡因这样的可卡因以粉末状盐的形式出现。在街头市场销售的可卡因通常会与各种粉末状填充剂混合,以增加其重量,从而使可卡因的成本更加昂贵。在这个过程中最常使用的物质是烘焙苏打;糖类,如乳糖和葡萄糖;以及局部麻醉剂,如利多卡因和苯佐卡因。苯佐卡因等物质会增强可卡因对粘膜的麻醉作用。总的来说,掺杂的可卡因通常是白色、象牙色或粉红色的粉末。
制备方法、杂质的存在以及可卡因的最初来源对于确定“裂解”可卡因的颜色非常重要。“裂解”可卡因的颜色通常在白色到淡黄奶油色到浅棕色之间变化。此外,它们的质地也会因切割可卡因的方式、粉末状可卡因的来源和加工以及转化为碱基的方法而有所不同。可卡因有时质地松脆,有时非常油腻,呈粉末状,几乎类似于晶体状。
可卡因有很多种形式。其中一种形式是盐。可卡因是一种弱碱性化合物,也称为“生物碱”,因此可以与酸性化合物结合形成各种盐。尽管硫酸盐 (-SO4) 和硝酸盐 (-NO3) 常见,但可卡因的盐酸盐 (HCl) 最为常见。不同性质的盐在各种溶剂中溶解和相互作用的方式不同。例如,盐酸盐 (HCl) 具有极性,并且在水中具有相当大的溶解度。可卡因的另一种形式是碱性。与盐的形式相反,“游离碱”一词指的是可卡因的碱性形式。与盐酸盐不同,任何可卡因碱性形式都不溶于水。因此,吸食游离碱可卡因的额外作用是由于物质的热解作用,导致甲基埃托根定释放到使用者体内,从而导致严重的肺组织和肝组织感染等问题。众所周知,通过液-液萃取技术,可以通过用碱性溶液中和其复合盐,然后沉淀为非极性碱性可卡因来制备纯可卡因。
此外,裂解可卡因是游离碱可卡因的低纯度形式。裂解可卡因通常通过用碳酸氢钠(碳酸氢钠,NaHCO3)和水的溶液中和盐酸可卡因来制备,最终产生一种非常粗糙/坚硬/易碎的、象牙色至棕色、无定形的物质,其中包含碳酸钠、截留的水和其 他副产物作为主要杂质。“游离碱”和“裂解”形式的可卡因易于使用,因为它们可以平滑地汽化。然而,盐酸可卡因不易汽化,因为它需要大约 197°C 的高温。最终,无论可卡因的形式如何,吸食或汽化可卡因并将其吸入肺部都会产生“兴奋”的感觉,并且随着可卡因的摄入,它会非常容易上瘾。
可卡因的兴奋感通常持续 10-30 分钟。可卡因在血液中传播并到达大脑的速度越快,兴奋感就越强烈。摄入可卡因后,使用者会体验到欣快感和精力充沛,包括心率和血压升高。可卡因的风险包括心脏病发作、中风、癫痫发作、腹痛、呼吸衰竭、恶心和死亡。由于使用者经常将可卡因与其他精神活性药物混合使用,因此猝死风险大大增加。可卡因通常与饮酒混合使用。研究人员发现,肝脏会将可卡因和酒精混合,产生可卡因乙酯,这会增强可卡因的欣快感,但也增加猝死的风险。可卡因会增加大脑中多巴胺的水平。多巴胺与快乐和运动有关。可卡因会阻止多巴胺被回收,导致神经递质积聚并不断放大信息。欣快感归因于多巴胺的不断积累。长期使用者会改变大脑的奖赏系统,导致可卡因滥用。可卡因会收缩血管,扩张瞳孔,增加心率和体温,还会引起头痛、腹痛和恶心。鼻内使用可卡因会导致嗅觉丧失、鼻出血和流鼻涕。瘾君子在两次回归之间经常会感到烦躁、不安和焦虑。幻觉和偏执也是常见的。
吸毒者使用可卡因,也称为裂缝或古柯作为娱乐性毒品。与大麻相比,可卡因是一种价格更高的致幻方式。可卡因也被称为“街头毒品中的鱼子酱”,以其神秘感而闻名,能使使用者保持警觉,并产生精力充沛的感觉。此外,吸毒者在可卡因的影响下,经常饮酒。使用者在感受到酒精影响很小的同时,感觉更加清醒和警觉。随着使用者更频繁地服用该药物,对该药物的耐受性会越来越高。由于可卡因会触发大脑的不同区域,并使使用者处于良好的兴奋状态,因此会导致上瘾。由于可卡因经常被使用,上瘾会导致身体和心理问题,甚至死亡。吸毒者可能会高度依赖这种毒品,当他们无法获得可卡因时,就会出现戒断症状。副作用可能包括疲劳、抑郁、焦虑、疼痛和酸痛。
参考文献
[edit | edit source]http://drugabuse.gov/drugpages/cocaine.html http://drugabuse.gov/infofacts/cocaine.html http://en.wikipedia.org/wiki/Cocaine#Suppository
"Cocaine Use." WEBMD. WEBMD LLC. Web. 18 Nov 2012. <http://www.webmd.com/mental-health/cocaine-use-and-its-effects>.
介绍
[edit | edit source]华法林是一种口服抗凝剂,用于预防新的血栓形成。血栓是在血管中血小板聚集和纤维蛋白网络形成的结果,该网络会捕获额外的血细胞,从而导致心脏病发作、中风或其他医疗问题。[1]
华法林还用于治疗静脉血栓形成(即静脉肿胀和血栓形成)。它用于预防肺栓塞(肺部血栓)。华法林也可以被称为抗凝剂(“血液稀释剂”)。
凝血
[edit | edit source]血液凝固是形成血栓的过程。它涉及一系列被称为“凝血级联”的过程。级联的每一步都涉及一种蛋白酶、一种酶原、一种非酶蛋白辅助因子、血小板表面和钙离子。在每一步中,酶原通过酶原中一个或多个肽键的断裂转化为活性蛋白酶。凝血酶是凝血酶原被蛋白水解激活的一系列凝血酶的最后一种。凝血酶然后将可溶性纤维蛋白原转化为不溶性纤维蛋白,最终形成纤维蛋白网状结构和最终的血栓。[2]
谁需要华法林?
[edit | edit source]患有人工心脏瓣膜、心律不齐、血栓形成和栓塞的患者被开出华法林。
概述
[edit | edit source]华法林的化学名称为 4-羟基-3-(3-氧代-1-苯基丁基)-2H-色烯-2-酮,化学式为 C19H16O4。
它的工作原理
[edit | edit source]华法林用于降低血液凝固的能力。它的作用机制是抑制维生素 K 依赖性凝血因子。它抑制凝血因子 II、VII、IX 和 X 的生成。这些因子需要谷氨酸残基的羧化才能与因子结合磷脂,这是一个与维生素 K 相关的过程。在维生素 K 被肝脏氧化成维生素 K 环氧化物后,维生素 K 环氧化物再被一种酶维生素 K 环氧化物还原酶 (VKOR) 还原成维生素 K。这种还原的环氧化物对于凝血因子的生成是必要的。因此,华法林在肝脏中起着抑制 VKOR 作用的作用,因此可以用于氧化和生成凝血因子的维生素 K 的可用量减少了。[3][4]
服用华法林的患者的饮食
[edit | edit source]建议避免摄入富含维生素 K 的食物,例如绿叶蔬菜、肝脏和植物油。
人们食用的食物会影响药物的作用方式。华法林是一种为有形成血栓高风险的人开出的药物。它可以预防有害的血栓形成,这些血栓可能会阻断流向心脏或大脑的血液。维生素 K 是一种通常存在于绿叶蔬菜中的维生素。与华法林相反,维生素 K 在人体血液凝固中起着重要作用。它用于逆转华法林等“血液稀释”药物的作用。华法林会降低维生素 K 的活性,并延长血栓形成所需的时间。因此,为了使华法林有效发挥作用,保持维生素 K 的摄入量尽可能一致非常重要。首先,保持高维生素 K 食物摄入量的稳定。其次,不要大幅度改变中等维生素 K 食物摄入量。第三,要注意膳食补充剂。服用维生素 E 或鱼油补充剂不明智。最后,营养师或医生可能会限制你的饮食中含有的蔓越莓汁。
| 食物 | 份量 | 维生素 K (mcg) |
|---|---|---|
| 羽衣甘蓝,煮熟 | 1/2 杯 | 531 |
| 菠菜,煮熟 | 1/2 杯 | 444 |
| 芥蓝,煮熟 | 1/2 杯 | 418 |
| 瑞士甜菜,生 | 1 杯 | 299 |
| 瑞士甜菜,煮熟 | 1/2 杯 | 287 |
| 芥菜,生 | 1 杯 | 279 |
| 芜菁甘蓝,煮熟 | 1/2 杯 | 265 |
| 欧芹,生 | 1/4 杯 | 246 |
| 西兰花,煮熟 | 1 杯 | 220 |
| 抱子甘蓝,煮熟 | 1 杯 | 219 |
| 芥菜,煮熟 | 1/2 杯 | 210 |
| 芜菁甘蓝,煮熟 | 1 杯 | 184 |
| 菠菜,生 | 1 杯 | 145 |
| 芜菁甘蓝,生 | 1 杯 | 138 |
| 菊苣,生 | 1 杯 | 116 |
| 西兰花,生 | 1 杯 | 89 |
| 卷心菜,煮熟 | 1/2 杯 | 82 |
| 绿叶生菜 | 1 杯 | 71 |
| 罗马生菜,生 | 1 杯 | 57 |
| 芦笋 | 4 根 | 48 |
副作用
[edit | edit source]副作用包括异常出血、腹泻、胸痛、面部肿胀、突然头痛或头晕、腿部剧烈疼痛、身体变色以及手指和脚趾变紫。
其他副作用包括味觉改变、疲劳、苍白、脱发,甚至感觉寒冷或发冷。华法林也会导致坏死或坏疽,这意味着皮肤或其他身体组织死亡)。[6]
参考文献
[edit | edit source]- ↑ Voet, Donald; Voet, Judith; Pratt, Charlotte (2006). Fundamentals of Biochemistry. Wiley.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ "Blood coagulation".
- ↑ Harrison, Karl (July 2004). "Warfarin".
- ↑ "Warfarin".
- ↑ "Nutrition care manual".
- ↑ American Society of Health-System Pharmacists (2012). "AHFS Consumer Medication Informatio".
布洛芬
[edit | edit source]
布洛芬通常用作非甾体抗炎药 (NSAID)。NSAID 是非甾体抗炎药的缩写。它用于缓解关节炎或发烧的症状。基本上,布洛芬起着止痛药或止痛药的作用。它通常存在于许多非处方药中,例如美林、泰诺、普渡和诺普林。换句话说,它通常以胶囊、片剂或粉末形式出现。与阿司匹林相比,布洛芬的作用时间较短,效果相对温和。然而,它已知具有抗血小板作用。布洛芬也被视为世界卫生组织 (WHO) 基本药物清单中的核心药物。这是一份基于特定医疗保健系统的最低医疗需求清单。
此外,布洛芬还起到血管收缩剂的作用,因为它抑制了由环氧合酶 2 酶产生的血管扩张前列环素。1969 年,布洛芬被称为一种羧酸,最初由英国的 Boot Pure Drug Company 以布鲁芬的名称发现。然而,许多人熟悉它是由安德鲁·R·M·邓洛普在约翰·尼科尔森和斯图尔特·亚当斯等人的帮助下发现的。即使在今天,布洛芬也以不同的形式作为止痛药出售,并以各种流行的商标(在非处方药中出售的品牌药物)出售。
通常,布洛芬用于治疗诸如疼痛、发烧甚至痛经之类的疾病。它还可以用于治疗类风湿性关节炎,这是一种已知的炎症性疾病。
布洛芬不仅有片剂或胶囊形式,还可以有局部凝胶形式,可以简单地通过皮肤吸收,这些凝胶通常用于运动损伤,因为它不会导致高风险的消化问题。
虽然布洛芬以其半衰期短而闻名,但它实际上有一个剂量依赖的时段,大约为 4 到 8 个小时。这相当长,特别是考虑到布洛芬的特性。没有人应该服用具体的剂量。换句话说,推荐剂量因人而异,根据他们的体重和适应症而异。即使没有具体的剂量,通常据说非处方使用的最大量大约为每剂 400 毫克,每天 1200 毫克。但是,对于那些耐受性和反应性更强的人来说有一个例外,因为他们可以服用的最大剂量是每剂 800 毫克,每天 3200 毫克。
布洛芬赖氨酸也称为布洛芬赖氨酸盐。有趣的是,布洛芬赖氨酸在欧洲、新西兰和澳大利亚等地被许可用于治疗与布洛芬相同的疾病。由于赖氨酸盐已知可以增加水溶性,因此它通常用于封闭早产儿(孕龄不超过 32 周,体重在 500 到 1500 克之间)的动脉导管。这恰好是一到三磅。如果布洛芬赖氨酸用于超过 32 周的婴儿,该药物将无效。布洛芬赖氨酸的使用是为了支持呼吸和限制液体。除了婴儿,布洛芬还可以用于帮助所有年龄段的人的肾脏功能。总的来说,与布洛芬酸相比,布洛芬赖氨酸显示出更快的作用,这证明了它的有效性。
布洛芬是由 Boot Pure Drug Company 发现的,其合成以该公司命名为“Boot 过程”。在该过程中,它从异丁苯的弗里德尔-克拉夫茨酰化开始。

“Boot 过程”
- 异丁苯与氯乙酸乙酯反应会生成α,β-环氧酯
- 水解
- 脱羧成醛
- 与羟胺反应生成肟
- 转化为腈,
- 水解成所需的酸

后来,以 Hoechst 公司命名的新的 Hoechst 过程进行了类似的乙酰化,然后用(雷尼镍)催化剂进行氢化。它会生成醇,最后一个反应是钯催化的羰基化。



布洛芬的功能是阻断一种名为环氧合酶 (COX) 的酶。NSAID 非选择性地抑制 COX-1 和 COX-2。当 COX 被抑制时,花生四烯酸被转化为 H2(PGH2),然后 PGH2 被转化为前列腺素和血栓烷 A2。前列腺素可以缓解炎症、发烧和疼痛,而血栓烷 A2 可以导致血栓形成。由于 COX-2 带来的唯一想要的功能(缓解疼痛、发烧和炎症)和 COX-1 会导致不想要的不良反应,因此人们对阻断 COX-2 但不阻断 COX-1 进行了进一步的研究。经过多年的研究,通过 X 射线晶体学和生物物理技术实现了这种选择性阻断。
布洛芬会导致某些人出现严重的过敏反应。如果出现以下症状,请立即寻求医疗帮助
- 荨麻疹
- 嘴唇、脸部、喉咙或舌头肿胀
- 呼吸困难
- 胸痛
- 视力问题
- 呕血
- 恶心
- 发烧
- 黄疸(皮肤或眼睛发黄)
- 抽搐
轻微的症状可能包括
- 胃部不适
- 腹泻
- 便秘
- 瘙痒
- 眩晕
- 耳鸣
布洛芬与某些其他药物混合使用会导致剧烈反应。这些药物包括但不限于
- 抗抑郁药
- 阿司匹林或其他 NSAID
- 心脏或血压药物
- 锂
- 利尿剂
- 类固醇
- 甲氨蝶呤
- 血液稀释剂
埃索美拉唑,商品名为耐信,被归类为一类称为质子泵抑制剂的药物。通过抑制胃壁细胞中的 ATPase,埃索美拉唑减少了胃酸的分泌量。该药物主要用于患有胃食管反流病 (GERD) 的人,这是一种胃酸过量导致胃酸反流并灼伤食道的疾病。通过阻止胃酸的形成,埃索美拉唑可以使受损的食道愈合并防止进一步的损伤。
埃索美拉唑的分子式为 (C17H18N3O3S)2Mg* 3 H2O,作为三水合物分子量为 767.2 克,无水物分子量为 713.1 克。
研究表明,接受高剂量埃索美拉唑的患者髋部、手腕或脊柱骨折的风险增加。高剂量定义为长期每天多次服用。此外,埃索美拉唑可能会导致严重的副作用,例如:• 水泡 • 荨麻疹 • 皮疹 • 瘙痒 • 呼吸或吞咽困难 • 心律不规则、快速或怦怦直跳 • 身体某部位不受控制的震颤 • 癫痫发作 • 胃痛 • 发烧
http://www.drugs.com/ibuprofen.html http://www.drugs.com/ibuprofen.html http://www.nlm.nih.gov/medlineplus/druginfo/meds/a682159.html http://www.news-medical.net/health/Ibuprofen-Mechanism.aspx http://www.chm.bris.ac.uk/motm/ibuprofen/welcome.htm http://en.wikipedia.org/wiki/Ibuprofen
(C22H23CIN2O2) 通常被称为开瑞坦,一种非处方药。它通常以白色至淡黄色粉末形式出现。它不溶于水,但极易溶于丙酮、酒精和氯仿。它被称为乙基 4-(8-氯-5,6-二氢-11H-苯并[5,6]环庚并[1,2-b]吡啶-11-亚甲基)-1-哌啶羧酸酯,分子量为 382.89 克。
片剂形式中,除了氯雷他定和抗组胺药外,还含有玉米淀粉、乳糖和硬脂酸镁等其他非活性成分。糖浆形式中,也含有氯雷他定和抗组胺药,但也包括其他非活性成分,例如柠檬酸、依地酸二钠、人工香料、甘油、丙二醇、苯甲酸钠和水。它的 pH 值约为 2.5-3.1。
氯雷他定用于缓解由灰尘、花粉和其他物质引起的过敏症状。氯雷他定属于抗组胺药类别,可以阻断 H1 受体上的组胺发挥作用,从而阻止组胺在体内引起过敏症状。常见的过敏症状包括但不限于打喷嚏、瘙痒和流鼻涕。氯雷他定是一种第二代组胺 H1 受体拮抗剂,因为它没有镇静作用。氯雷他定有 4 种不同的形式:液体、口服混悬液、立即释放或缓释片剂以及口崩片。氯雷他定还可以与伪麻黄碱联合使用。口服后,氯雷他定通过胃肠道吸收,并被细胞色素 P450 代谢,然后与血浆膜蛋白结合。
虽然氯雷他定被认为是“非镇静性”抗组胺药,但已注意到剂量相关的镇静作用。因此,在与其他中枢神经系统抑制剂(如巴比妥类药物、苯二氮卓类药物、阿片类激动剂、抗精神病药、三环类抗抑郁药、乙醇、其他 H1 受体阻滞剂以及抗焦虑药、镇静剂和催眠药)联合使用时,务必注意嗜睡症状。


体外研究表明,氯雷他定主要由 CYP3A4 代谢为去碳乙酰基氯雷他定,部分由细胞色素 CYP2D6 代谢。CYP3A4 抑制剂,氯雷他定主要由 CYP2D6 代谢为去碳乙酰基氯雷他定。酮康唑、红霉素(均为 CYP3A4 抑制剂)或西咪替丁(CYP2D6 和 CYP3A4 抑制剂)会增加氯雷他定的血浆浓度。氯雷他定的半衰期约为 8.4 小时,其代谢产物的半衰期约为 28 小时。消除通过粪便和肾脏途径进行。
西咪替丁、红霉素、奈法唑酮和酮康唑已被证明会干扰氯雷他定的代谢。P-450 CYP3A4 同工酶的抑制导致氯雷他定及其代谢产物的血清浓度升高。与对照组患者相比,氯雷他定血清浓度升高不会导致不良反应的显着差异。然而,在氯雷他定与其他药物联合使用时,需要注意心律失常事件的并发风险。虽然尚未确定氯雷他定与细胞色素 P-450 之间的药物相互作用,但由于这些药物与某些其他 H1 受体拮抗剂之间的相互作用的严重性,需要注意强效 CYP3A4。
有些人服用这种药物后会出现过敏反应。一些严重的副作用包括
- 心跳快速且不规律
- 黄疸(皮肤或眼睛发黄)
- 癫痫发作
- 感觉快要晕倒
有些人可能出现轻微的过敏反应,表现出以下症状,但没有那么严重
- 头痛
- 恶心
- 紧张
- 嗜睡
- 胃痛、腹泻
- 口干
- 喉咙痛
建议出现任何严重症状的人立即联系医生。
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001010/ http://en.wikipedia.org/wiki/Loratadine http://www.rxlist.com/claritin-drug.htm http://www.rxlist.com/claritin-drug.htm
埃索美拉唑,商品名为耐信,被归类为一类称为质子泵抑制剂的药物。通过抑制胃壁细胞中的 ATPase,埃索美拉唑减少了胃酸的分泌量。该药物主要用于患有胃食管反流病 (GERD) 的人,这是一种胃酸过量导致胃酸反流并灼伤食道的疾病。通过阻止胃酸的形成,埃索美拉唑可以使受损的食道愈合并防止进一步的损伤。
埃索美拉唑的分子式为 (C17H18N3O3S)2Mg* 3 H2O,结构式如下所示。

单次口服 20-40 毫克剂量通常在 1-4 小时内产生 0.5-1.0 毫克/升的埃索美拉唑血浆峰值浓度,但在连续数天每日一次给药后,这些水平可能会增加约 50%。类似剂量的 30 分钟静脉输液通常会产生 1-3 毫克/升的血浆峰值水平。该药物从体内快速清除,主要通过尿液排泄药理学上无活性代谢产物,如 5-羟甲基埃索美拉唑和 5-羧基埃索美拉唑。除非采用手性技术,否则埃索美拉唑及其代谢产物在分析上与奥美拉唑和相应的奥美拉唑代谢产物无法区分。
研究表明,接受高剂量埃索美拉唑的患者髋骨、腕骨或脊柱骨折风险增加。高剂量定义为长期每日多次服用。此外,埃索美拉唑可能会引起以下严重副作用:
• 水泡
• 荨麻疹
• 皮疹
• 瘙痒
• 呼吸或吞咽困难
• 心跳不规则、快速或怦怦直跳
• 身体某一部位无法控制的颤抖
• 癫痫发作
• 胃痛
• 发烧 甲氨蝶呤

一种用于治疗癌症患者的化疗药物,具有免疫抑制特性。它是二氢叶酸还原酶的抑制剂,有助于 RNA 的形成。它还抑制胸苷酸合成酶,胸苷酸合成酶是合成 DNA 所必需的。

甲氨蝶呤用于治疗乳腺癌、头颈癌、肺癌、胃癌和食道癌、急性淋巴细胞白血病 (ALL)、非霍奇金淋巴瘤 (NHL)、妊娠滋养细胞肿瘤和蕈样肉芽肿 (皮肤 T 细胞淋巴瘤)。甲氨蝶呤的给药方式为
- 静脉输液
- 肌肉注射
- 另一种给药方式是脑室或鞘内输液,直接注入脊髓液。
- 口服形式
- 血细胞计数低
- 口腔溃疡
- 恶心和呕吐
- 肾毒性
- 皮疹、皮肤发红
- 腹泻
- 脱发
- 眼睛刺激
- 生育能力下降

癌细胞在其细胞分裂方面不同于正常细胞。当正常细胞接触到与它们相同的细胞时,它们会停止分裂。这种机制被称为接触抑制。癌细胞没有这种能力。癌细胞的细胞分裂没有限制,导致肿瘤形成。与正常细胞类似,癌细胞也经历细胞周期,包括静止期、活跃生长期和有丝分裂。
化疗能够通过破坏负责细胞分裂的 DNA 或 RNA 来杀死癌细胞。如果癌细胞不能分裂,肿瘤就会缩小。化疗药物对正在分裂的细胞有效,被称为细胞周期特异性;或对处于静止状态的细胞有效,被称为细胞周期非特异性。化疗方案的制定基于癌细胞类型及其分裂速度。
然而,化疗药物无法区分癌细胞和正常细胞,从而导致副作用。化疗最常影响的正常细胞是血细胞、口腔细胞、胃细胞和毛囊细胞;导致血细胞计数低、口腔溃疡、恶心、腹泻和/或脱发。
甲氨蝶呤被归类为抗代谢药。抗代谢药与正常的细胞物质非常相似。当抗代谢药进入细胞代谢时,它们会与正常的细胞物质竞争,抑制某些途径,最终抑制细胞分裂。甲氨蝶呤通过与癌细胞中的叶酸竞争而发挥其化疗作用,导致细胞中叶酸缺乏,最终导致细胞死亡。甲氨蝶呤还会与正常细胞中的叶酸竞争,并导致严重的副作用,例如血细胞计数降低、脱发、口腔溃疡、腹泻、肝脏、肺脏、神经和肾脏损伤。
甲氨蝶呤的化疗作用是由于它能够通过抑制二氢叶酸还原酶和胸腺嘧啶合成酶来限制 DNA 和 RNA 的合成。它通过叶酸使用的主动转运系统进入细胞。然后甲氨蝶呤会进行多聚谷氨酰化。MTXGlu在抑制酶方面更有效。二氢叶酸还原酶通过还原二氢叶酸(在胸腺嘧啶合成过程中产生)来维持还原叶酸的水平。二氢叶酸还原酶将叶酸还原为四氢叶酸,它是嘌呤核苷酸合成中必需的辅因子。
"甲氨蝶呤"。Scott Hamilton CARES Initiative。N.p.. 网页。2012 年 11 月 21 日。<http://chemocare.com/chemotherapy/drug-info/Methotrexate.aspx>
Tian, Henghe,和 Bruce N. Cronstein。"纽约大学医院关节疾病公报"。纽约大学医院关节疾病公报。65.3 (2007): 168-73. 印刷。
Brigitte C. Widemanna, Peter C. Adamson b. 了解和管理甲氨蝶呤肾毒性儿科肿瘤学 2006;11;694-703。

万古霉素被认为是最后的抗生素,并且往往对许多(如果不是所有)其他药物高度耐药的细菌有效。它是一种糖肽,来源于东方链霉菌培养物。万古霉素最有效的细菌之一是耐甲氧西林金黄色葡萄球菌(MRSA)。总体而言,万古霉素对各种革兰氏阳性细菌有效,包括高度耐药的链球菌、肠球菌和葡萄球菌菌株。万古霉素不应作为口服药物服用,因为它不能通过这种方式吸收,而应通过静脉注射。 [1]
万古霉素通过与细胞壁中肽聚糖链的末端氨基酸结合起作用。通过这样做,通过不允许多肽聚糖链之间发生交联,可以阻止细菌细胞壁的生成。如果没有坚固的细胞壁,就会诱导细胞裂解,细菌细胞会崩解。 [1]
在细菌细胞中,细胞壁由肽聚糖组成。这种肽聚糖是由两个糖分子重复单元组成的,即 N-乙酰葡萄糖胺(NAG)和 N-乙酰胞壁酸(NAM)。与 NAM 相连的是一系列以 D-丙氨酸二肽结束的氨基酸。在细胞膜的细胞质内,NAM 和 NAG 在细菌萜醇处连接形成二糖 NAM-NAG。NAM-NAG 被细菌萜醇带到细胞膜的外部区域,青霉素结合蛋白 (PBP) 与 D-Ala 二肽结合。一个 PBP 是转肽酶,它将两个 NAG 分子的肽侧链连接在一起形成肽交联。这种肽交联通过连接一个 NAM 侧链的 L-赖氨酸到另一个 NAM 侧链的 D-Ala 来帮助增加细胞壁的稳定性。 [2]
万古霉素与 NAM-NAG 二糖的 D-Ala-D-Ala 末端结合,阻止转肽酶形成肽交联。 [2] 由于自溶素继续断裂肽交联,并且由于万古霉素的存在,无法形成新的交联,因此细菌由于渗透裂解而破裂。
盐酸万古霉素主要用于治疗由对耐甲氧西林葡萄球菌敏感菌株引起的严重感染。通常,万古霉素是一种最后的手段抗生素,通常用于对青霉素过敏的患者、对其他类型的抗生素(如头孢菌素)没有反应的患者以及受万古霉素敏感细菌困扰的患者。一旦确定细菌类型并获得敏感性数据,就应确定治疗方案。它对葡萄球菌性心内膜炎、其他葡萄球菌感染(骨感染、呼吸道感染和皮肤感染)有效。万古霉素对其他类型的感染有效,包括白喉杆菌性心内膜炎。为了提高对抗细菌感染的有效性,万古霉素通常与氨基糖苷类药物或利福平一起使用。万古霉素对病毒感染无效。
与所有药物一样,每种药物通常都存在风险和副作用。与大多数抗生素一样,耐药菌的发生风险已成为一场日益加剧的战斗。当耐药菌形成时,治疗和对抗与这些细菌相关的感染变得更加困难。万古霉素必须静脉注射,因为该药物对组织有很强的刺激性。如果药物与肌肉组织相互作用,通常会导致肌肉疼痛、压痛,并且组织开始死亡,因此支持静脉注射万古霉素的必要性。此外,长期使用万古霉素与其他药物一起可能会产生不良反应。例如,与麻醉剂的长期使用可能会导致低血压、瘙痒和红斑等并发症。抗生素的一些副作用通常包括腹泻和胃痛。其他影响包括过敏反应、药物发烧、恶心、寒战和皮疹。
来源:http://www.drugs.com/pro/vancomycin-hydrochloride.html
万古霉素会在某些人身上引起剧烈的过敏反应。最常见的严重症状是
- 荨麻疹
- 嘴唇、面部、喉咙或舌头肿胀
- 听力损失
- 发烧
- 头晕
- 剧烈的胃痛
- 昏厥
与某些其他药物混合使用时,万古霉素会导致严重反应。与万古霉素反应最常见的药物是
- 阿米卡星
- 庆大霉素
- 卡那霉素
- 新霉素
- 链霉素
- 妥布霉素
参考资料:http://www.drugs.com/vancomycin.html http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000282/ http://pharmacologycorner.com/vancomycin-mechanism-action-animation/
- ↑ a b c Dewick, Paul M. 药用天然产物:生物合成方法,第三版,John Wiley and Sons Ltd. 2009 年。
- ↑ a b Slonczewski, Joan L. Foster, John W. 微生物学:不断发展的科学,第二版,W.W. Norton & Company. 2009 年。
反应停于1953年由西德一家公司合成,用作镇静剂。它在20世纪50年代中期在美国以外地区流行,用于缓解孕吐,但在1961年被发现会导致出生缺陷。它被停止广泛使用,但现在被用于治疗其他疾病,如麻风病和多发性骨髓瘤。 [1]
成分
[edit | edit source]反应停是一种无味无臭的白色结晶化合物。 [2]
反应停的化学式为C13H10N2O4,以外消旋混合物的形式存在。
合成
[edit | edit source]反应停的合成方法如下所示:两步合成反应不需要任何纯化步骤。L-谷氨酰胺与N-羧乙氧基邻苯二甲酰亚胺反应生成N-邻苯二甲酰基-L-谷氨酰胺。在催化量的DMAP存在下,用CDI处理N-邻苯二甲酰基-L-谷氨酰胺,使其环化生成反应停。 [3] [3]
医学重要性
[edit | edit source]尽管反应停与广泛的悲剧事件有关,但由于它在治疗麻风病和癌症等多种疾病方面具有治疗作用,因此它仍在医疗环境中使用。
反应停在治疗麻风病和多发性骨髓瘤等疾病中最有效的方面是它能够抑制血管生成并具有抗炎作用。在麻风病中,反应停在抑制与之相关的疼痛方面特别有效。
[4]
在一项针对晚期多发性骨髓瘤患者的研究中,反应停被证明能够使10%的患者缓解。这可能是由于多种因素造成的,包括抑制血管生成、反应停改变粘附分子表达的能力,以及它对肿瘤坏死因子-α生长的抑制作用。 [5]
反应停通过降解其mRNA选择性地抑制肿瘤坏死因子-α,从而降低该分子的半衰期。 [6] 抑制这种因子(它会导致癌症患者发烧和不适)可以极大地提高生活质量。然而,它也可能削弱免疫系统。 [span>7]
反应停还可以帮助缓解HIV引起的消瘦,因为它能抑制肿瘤坏死因子-α。 [span>4].
致畸作用
[edit | edit source]母亲在怀孕期间服用反应停的孩子,通常出生时四肢短小或缺失。
反应停的结构导致了它的致畸作用。测试表明,只有反应停的S对映异构体具有致畸性。反应停最初是外消旋的,但不能通过分离一种对映异构体来使其安全,因为R对映异构体在体内很容易转化为S对映异构体。 [8]
关于反应停的作用机制,尤其是与致畸性相关的机制,已经提出了各种假设。然而,目前的研究主要集中在“(1)氧化应激/损伤,(2)DNA嵌入,(3)抑制血管生成,以及(4)与芹菜素(CRBN)结合”。 [span>4].
- 研究表明,反应停可以促进给予它的老鼠产生氧自由基,导致氧化应激,因为生物可利用的活性氧减少,影响了大量依赖氧的器官。
- 反应停通过DNA嵌入附着于富含GC的启动子位点,降低IGF-1和FGF-2的转录,两者都导致血管生成以及四肢和肢体的正常生长。
- 反应停是一种血管生成抑制剂,这意味着它可以阻止新血管的形成。这对治疗糖尿病性视网膜病变、黄斑变性和实体瘤很有用。 [9]
- 研究发现,CRBN是一种与反应停结合的蛋白质,它的突变体与反应停的结合能力较差,当过度表达时,可能会削弱反应停的致畸性肢体影响。
这些机制可能以某种方式共同导致了反应停的整体致畸作用。
当给怀孕的兔子(对反应停敏感)和怀孕的大鼠(对反应停不敏感)服用反应停时,兔子胚胎肢芽细胞中的绿色荧光蛋白表达减少,而大鼠则没有受到影响。 [4].
暴露时间与反应停引起的特定的问题有关:http://toxsci.oxfordjournals.org/content/122/1/1/F3.large.jpg
药物试验
[edit | edit source]反应停最初进行了动物测试以及人体临床试验。这包括98名1岁以下的儿童、160名哺乳期妇女和81名怀孕第三三周的妇女。在临床试验期间没有观察到致畸作用。 [2] 反应停导致数千例出生缺陷后,在孕妇身上测试药物以检测致畸作用成为一项强制性规定。 [1]
参考文献
[edit | edit source]- ↑ a b [2]
- ↑ a b The Saga of Thalidomide
- ↑ a b c A Concise Two-Step Synthesis of Thalidomide Invalid
<ref>tag; name "A Concise Two-Step Synthesis of Thalidomide" defined multiple times with different content - ↑ a b c d Thalidomide: The Tragedy of Birth Defects and the Effective Treatment of Disease
- ↑ Antitumor Activity of Thalidomide in Refractory Multiple Myeloma
- ↑ Moreira et. al.
- ↑ Sampaio et. al
- ↑ A Concise Two-Step Synthesis of Thalidomide
- ↑ Angiogenesis Inhibition
概述
[edit | edit source]辛伐他汀的实验式为C25H38O5,分子量为418.57。辛伐他汀也是一种白色结晶粉末,不溶于水。服用辛伐他汀时,要确保我们不过敏。服用辛伐他汀的患者必须限制酒精饮料的摄入。
辛伐他汀,也称为佐寇,是一种用于控制胆固醇水平的药物。它是一种降脂药,用于降低脂类水平。它源于土曲霉的发酵,土曲霉是一种可以用于生产重要有机化合物的真菌。20世纪50年代初,默克公司的生物化学家杰西·哈夫及其同事研究了胆固醇的生物合成。1956年,默克公司的卡尔·霍夫曼、卡尔·福克尔斯及其同事从酵母提取物中分离出甲羟戊酸。他们证实这种酸可以用于胆固醇的生物合成。1976年,日本三共制药的远藤章能够从桔青霉中分离出紧密霉素,桔青霉是一种真菌。1979年,卡尔·霍夫曼能够从土曲霉中分离出洛伐他汀。默克公司的科学家们从洛伐他汀中合成了辛伐他汀。
严格按照医嘱服用西美伐他汀。切勿过量服用,或服用时间超过医生建议的时间。请仔细遵照医生的剂量说明。过量服用此药可能会导致严重或危及生命的副作用。
西美伐他汀通常在睡前或晚餐时服用。如果您每天服用西美伐他汀不止一次,请在餐后服用。医生可能会偶尔调整您的剂量,以确保您获得最佳效果。为了确保西美伐他汀对您的病情有帮助,并且没有造成有害的影响,需要定期进行血液检查。您的肝功能应该每 3 到 6 个月检查一次。定期去看医生。您可能需要长期服用西美伐他汀来治疗高胆固醇。如果您要进行手术或发生医疗紧急情况,您可能需要短期内停止使用此药。除非医生指示,否则不要停止服用西美伐他汀。
西美伐他汀只是完整治疗方案的一部分,该方案还包括饮食、运动和体重控制。
每天饮酒可能会增加您患肝脏问题的风险。其他副作用包括无法解释的肌肉疼痛、压痛、无力;发烧、异常疲倦和深色尿液;排尿时疼痛或灼痛;恶心、上腹部疼痛、瘙痒、食欲不振或深色尿液。[内部药物索引]。
西美伐他汀含有某些非活性成分,如果您过敏,可能会引起过敏反应。如果患者有饮酒史、肾脏疾病或肝脏疾病,应谨慎使用。如果需要进行手术,请告知医生您使用过的草药、非处方药和处方药。服用西美伐他汀时,请注意您的饮酒量。与酒精一起服用,这种药物会增加肝脏问题的风险。老年人应更加谨慎。他们可能对这种药物更加敏感,可能会导致肌肉问题。怀孕期间不要使用这种药物,因为它会伤害您的未出生婴儿。此外,预防怀孕也很重要,因为西美伐他汀可能会进入母乳。尽管这一点尚不清楚,但在服用这种药物期间,尽量不要母乳喂养。
很少有服用过这种药物的人报告过轻微记忆力减退或意识模糊的问题。这些是可能发生的罕见副作用。不常见的是,可能会发生肌肉问题,并且可能发生非常严重的状况,横纹肌溶解症(肌肉纤维分解,导致血液中出现肌肉纤维。请注意包括虚弱、压痛或肌肉疼痛在内的症状。在极少数情况下,可能会发生肝脏问题。请注意恶心或呕吐、严重的腹部或胃痛、深色尿液或皮肤或眼睛发黄。这种药物也可能导致严重的过敏反应。请注意呼吸困难、严重头晕、面部、喉咙或舌头发痒或肿胀、皮疹或任何过敏反应的症状。
1)在服用大麻之前,抑制性神经递质在突触中是活跃的。这些神经递质的作用是抑制多巴胺释放到突触中。

2)当被体内天然大麻素激活时,大麻素受体关闭抑制性神经递质的释放。当没有抑制时,多巴胺被释放。

3)四氢大麻酚(THC)是大麻中的活性化学物质,它模拟anandamide并与大麻素受体结合。抑制被关闭,多巴胺开始进入突触。

4)anandamide以参与消除短期记忆而闻名。它还负责产生放松和平静的感觉,并减缓运动速度。与 THC 不同,anandamide 非常脆弱,并且在体内很容易分解。这就是为什么anandamide 不会产生永久的自然“兴奋”的原因。
1)多巴胺完成其工作后,被多巴胺转运分子从突触间隙中取出。

2)可卡因的作用是阻断转运体,使多巴胺停留在突触间隙中。结果,多巴胺不断地与受体结合,导致细胞过度刺激。

3)最后,可卡因集中在大脑的奖励通路中。它还激活了大脑中控制随意运动的部分。这就是为什么滥用可卡因的人无法保持静止的原因。
1)GABA 是一种抑制性神经递质,在整个大脑中都是活跃的。这些神经递质控制着许多脑通路中的神经活动。当 GABA 与其受体结合时,细胞不太可能放电。

2)在大脑的另一个区域,谷氨酸充当大脑的通用兴奋性神经递质。
 3)酒精进入大脑后,会产生镇静作用。它与 GABA 受体相互作用,并驱动它们变得更加抑制。然后,它与谷氨酸受体结合,抑制谷氨酸神经递质与其受体结合。结果,细胞被抑制兴奋。
3)酒精进入大脑后,会产生镇静作用。它与 GABA 受体相互作用,并驱动它们变得更加抑制。然后,它与谷氨酸受体结合,抑制谷氨酸神经递质与其受体结合。结果,细胞被抑制兴奋。
 4)因此,酒精会影响大脑中负责记忆形成和决策的区域(额叶)。
4)因此,酒精会影响大脑中负责记忆形成和决策的区域(额叶)。
1)在突触中活跃的抑制性神经递质抑制多巴胺神经递质的释放。

2)当由身体产生的天然阿片类物质(内源性阿片类物质)激活阿片类受体时,抑制性神经递质的释放被关闭。当没有抑制时,多巴胺被释放,并与多巴胺受体结合。

3)当海洛因进入系统时,它会与阿片类受体结合,因为它与内源性阿片类物质具有相同的形状。这种机制关闭了多巴胺的抑制,多巴胺被释放到突触中。这反过来会产生镇静和幸福的感觉。

4)这些阿片类受体与负责疼痛传递、应激反应和情感依恋的神经元相连。因此,类似海洛因的药物被用作止痛药,以减轻严重损伤的疼痛。
1)LSD 药物只对一种称为血清素的神经元起作用。LSD 的形状与血清素相似,并通过与受体结合来减弱血清素的作用。

2)大脑中有许多种血清素受体,每种受体负责特定的功能。

3)LSD 与某些受体结合,但方式不同。LSD 可以抑制或兴奋这些受体。这就是 LSD 具有复杂感觉效应的原因。

4)LSD 与其他致幻剂一样,会兴奋大脑中一个称为蓝斑的特定区域。在这个区域,有一个神经元与其相连,该神经元分支到大脑中的不同感觉区域。蓝斑负责清醒的感觉和对意外刺激的非自主反应。
1)多巴胺通过多巴胺转运体从突触间隙中移除。甲基苯丙胺被摄入细胞,因为甲基苯丙胺模拟多巴胺的形状,而多巴胺转运体无法识别多巴胺与甲基苯丙胺之间的区别。

2)当甲基苯丙胺被转运到细胞内部时,它会进入携带多巴胺的囊泡,并迫使多巴胺从囊泡中排出。

3)当细胞内部的多巴胺含量丰富时,多巴胺通过多巴胺转运体的逆向泵送,被泵出细胞,进入突触间隙。

4) 多巴胺一旦离开细胞,就会反复与多巴胺受体结合,因为它在突触间隙中非常丰富。

5) 冰毒直接作用于大脑的奖励通路。因此,它具有很强的成瘾性,冰毒使用者在服用后会感到强烈的快感。
摇头丸
[edit | edit source]1) 血清素完成其工作后,会通过血清素转运蛋白从突触间隙中移除。

2) 摇头丸会模仿血清素的形状,被血清素转运蛋白吸收。实际上,血清素转运蛋白对摇头丸的亲和力比对血清素本身的亲和力更高。

3) 血清素转运蛋白与摇头丸的相互作用会使转运蛋白混乱,导致它们开始将血清素从细胞中泵入突触间隙。

4) 由于血清素在突触间隙中变得丰富,它会开始反复与受体结合,导致细胞过度刺激。

5) 摇头丸会影响睡眠、情绪、感知和食欲。它还会间接影响奖励通路。摇头丸的成瘾性不如其他药物,因为它沿奖励通路释放的多巴胺较少。
赫勒综合征
[edit | edit source]赫勒综合征是一种遗传性疾病,与皮肤硫酸软骨素和肝素硫酸的储存和尿液排泄有关。患有赫勒综合征的人由于α-L-艾杜糖醛酸酶缺乏症,无法分解称为粘多糖或糖胺聚糖(GAGs)的长糖分子,α-L-艾杜糖醛酸酶是一种负责降解糖胺聚糖的溶酶体酶。GAGs 会积聚在脑、心脏、肝脏、骨骼和其他器官的溶酶体中。GAGs 在人体内的积累会导致器官损伤。
症状
[edit | edit source]- 骨骼异常
- 角膜混浊
- 耳聋
- 身材矮小
- 心脏病
- 关节疾病,包括僵硬
- 智力障碍
- 面部特征粗糙,例如鼻梁低平、脸扁平、头大
- 肝脾肿大
- 腹泻
- 呼吸困难和打鼾
赫勒综合征患儿出生时看起来正常。症状开始出现在 6 个月到 2 岁之间,最常出现在 3 岁到 8 岁之间。大多数患有赫勒综合征的人无法活过 10 岁。
背景信息
[edit | edit source]赫勒综合征最初是由一位名叫伊丽莎白·F·纽菲尔德的研究人员在 20 世纪 60 年代后期提出的。最初,纽菲尔德将该疾病的原因与硫酸软骨素 B 和肝素硫酸的聚合物联系起来,但由于它们的化学结构不同,很难确定缺陷。她后来假设粘多糖链的过度合成以及 UDP-葡萄糖醛酸的过量,是由 UDP-木糖反应缺陷造成的,这是导致这种情况的原因。然而,当抑制 UDP-葡萄糖脱氢酶时,她建立的实验性细胞培养中并没有产生任何影响。当她将(35)SO4 的放射性应用于具有高溶解度的生物大分子以测量粘多糖的含量时,她的假设再次被证明是错误的。赫勒患者的细胞中粘多糖含量没有下降,而正常人的样本则下降了。通过这两个实验,纽菲尔德后来得出结论,赫勒综合征是一种溶酶体贮积病,是由无法分解粘多糖引起的。
治疗
[edit | edit source]已经开发出一种称为酶替代疗法的治疗方法来解决赫勒综合征。使用人类和犬类的α-L-艾杜糖醛酸酶 cDNA 在中国仓鼠卵巢细胞中生产重组酶。这些细胞分泌带有 6-磷酸甘露糖信号的α-L-艾杜糖醛酸酶。6-磷酸甘露糖是一种碳水化合物修饰,它会抑制α-L-艾杜糖醛酸酶的摄取。这种重组酶在α-L-艾杜糖醛酸酶缺乏症的犬类中进行了测试。它在短期和长期实验中都降解了许多器官中的 GAGs。这种重组酶后来在人体上进行了测试,并于 2003 年获得 FDA 批准。
虽然已被证明有效,但酶替代治疗也有一些局限性。并非所有组织都能得到同样程度的矫正1。由于血脑屏障的存在,储存在中枢神经系统中的粘多糖无法被酶所及。最后,酶替代治疗非常昂贵,每年需要 50 万美元1。
赫勒综合征的另一种治疗方法是造血干细胞移植。这种方法有助于维持正常的智力发育。该治疗方法只有在生命早期进行才有效。这种治疗方法的另一个局限性是它存在风险。
参考文献
[edit | edit source]- Neufeld, Elizabeth F. (2011). "从意外发现到治疗". 生物化学年度综述. 80: 1–15. doi:10.1146/annurev.biochem.031209.093756. PMID 21675915.
- Kakkis, E.D.; Matynia, A.; Jonas, A.J.; Neufeld, E.F. (1994). "人溶酶体酶α-L-艾杜糖醛酸酶在中国仓鼠卵巢细胞中的过表达". 蛋白质表达与纯化. 5 (3): 225–32. doi:10.1006/prep.1994.1035. PMID 7950365.
- http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002184/
- Scott, H. S.; Ashton, L. J.; Eyre, H. J.; Baker, E; Brooks, D. A.; Callen, D. F.; Sutherland, G. R.; Morris, C. P.; Hopwood, J. J. (1990). "人α-L-艾杜糖醛酸酶基因(IDUA)的染色体定位到 4p16.3". 美国人类遗传学杂志. 47 (5): 802–7. PMC 1683689. PMID 2220820.
- Stoltzfus, L. J.; Sosa-Pineda, B; Moskowitz, S. M.; Menon, K. P.; Dlott, B; Hooper, L; Teplow, D. B.; Shull, R. M.; Neufeld, E. F. (1992). "犬α-L-艾杜糖醛酸酶 cDNA 的克隆和鉴定。粘多糖代谢症 I 犬的 mRNA 缺陷". 生物化学杂志. 267 (10): 6570–5. PMID 1551868.
- http://medschool.ucsf.edu/lysosomal/hurler/
基于 RNA 的药物
[edit | edit source]RNAi 药物是一种相对较新的药物类别,它依赖于一种称为 RNA 干扰的机制来抑制或沉默某些基因的基因表达。具体来说,RNAi 药物由合成的短干扰 RNA 分子 (siRNA) 制成,这些分子会招募并抑制编码特定基因的特定 mRNA 转录本的表达[3]。因此,RNAi 药物治疗癌症背后的策略是,通过合成与引起癌症基因的特定 mRNA 结合的 siRNA 分子,siRNA 通过 RNA 干扰机制可以使编码有害蛋白质的 mRNA 转录本失活,从而抑制该蛋白质的翻译,而该蛋白质会导致癌症和疾病的症状。由于 siRNA 能够降解 mRNA 并沉默其基因,因此基于 RNAi 的药物被认为是治疗癌症的一种新的创新方法。基于 RNA 的治疗方法的可能适应症包括 HIV、黄斑变性以及治疗癌性肿瘤细胞。
全身 siRNA 递送策略可以分为被动递送和主动(靶向)递送。
自 2002 年发现 RNA 干扰以来,科学家们一直在积极研究 RNA 干扰抑制导致各种人类疾病和癌症的有害或癌性基因表达的潜力。例如,科学家们开始利用 RNA 干扰来治疗黄斑变性,这是一种由眼睛中 VEGF 基因过度表达引起的疾病,导致视网膜后面血管过度生长,最终会导致失明。[1] 因此,科学家们最初寻求将 RNAi 药物用作关闭眼睛中过度活跃的 VEGF 基因的方法。
RNAi 的另一个潜在用途一直在积极研究中,用于治疗 HIV,并正在导致一类新型 HIV 药物的开发。2011 年 1 月发表的一项研究发现,将与特异性结合 HIV 细胞表面的 RNA 适体分子结合的 siRNA 分子递送,显着降低了注射病毒的小鼠的 HIV 病毒表达。[4]。
由于 RNAi 的药物递送系统存在困难,因此 RNAi 药物的生产和营销一直特别具有挑战性;RNA 在人体血液中被降解,这使得药物难以到达人类细胞中的预期作用部位以发挥作用。[2] 为了解决这个问题,科学家们正在研究将 RNAi 分子与其他分子偶联的方法,以防止 RNAi 分子在到达细胞之前在血液中过早降解,或者将 RNAi 分子包装在封闭的分子中,这些分子可以运输 RNAi 分子以确保其有效递送。例如,加州大学旧金山分校的研究人员正在探索将 siRNA 包装在称为脂质体的脂肪涂层分子中的方法,作为一种药物递送方式,脂质体已广泛用作商业药物开发中的药物载体。[3] 一旦被运输到预期的作用部位,siRNA 含量就可以从脂质体中释放出来,以沉默该部位的基因。
- Lewis, Susan K. “The RNAi Cure?” PBS. 1 July 2005 <http://www.pbs.org/wgbh/nova/body/rnai-cure.html>.
- Pollack, Andrew. “Drug-Makers Fever for the Power of RNA Interference Has Cooled”. The New York Times. 7 February 2011 <http://www.nytimes.com/2011/02/08/science/08rna.html?_r=1&pagewanted=all>.
- Norris, Jeffrey. “UCSF Leads Consortium to Radically Change Cancer Drug Development”. UCSF. 21 July 2009 <http://www.ucsf.edu/news/2009/07/8192/sirna-drug-development-university-labs>
- Willyard, Cassandra. “Double Whammy for HIV”. Nature. 19 January 2011 < http://www.nature.com/news/2011/110119/full/news.2011.30.html>.

抗生素于 1928 年首次用青霉素生产,非常有效。许多制药公司以青霉素为榜样,后来自己生产了其他抗生素产品。抗生素对人类、动物和活细菌迅速产生了重大影响。根据一篇名为“细菌的多重耐药性”的文章,由 Hiroshi NIkaido 撰写,估计每年生产 100,000 吨抗生素;由于其治愈疾病的能力和受欢迎程度,我们利用并过度使用抗生素;这导致了多重耐药性。
抗生素在临床环境中的使用一直受抗生素的一些关键特征的指导。
- 抗生素具有选择性毒性:这使得能够检测细菌靶标而不是人体所需的真核细胞。
- 抗生素具有有限的活性范围:因此每种抗生素只影响少数细菌种类,因此需要许多不同的抗生素。例如青霉素,它主要杀死革兰氏阳性细菌。另一方面,氨苄青霉素具有一个额外的氨基,允许穿透革兰氏阴性细菌的外膜。因此,氨苄青霉素比青霉素具有更广泛的活性范围。
- 抗生素可以是杀菌剂或抑菌剂。
- 杀菌剂抗生素杀死细菌微生物。
- 抑菌剂抗生素阻止细菌生长。[1]
许多抗生素会阻止蛋白质合成。所有细胞生物体,包括细菌,都具有核糖体。所有核糖体都由蛋白质和核糖体 RNA 组成。但是,这些蛋白质的精确形状在人类和细菌之间有几种非常特定的差异。这对试图开发称为抗生素的杀菌药物的研究人员来说是一件好事,因为它意味着科学家们可以设计出能够破坏细菌核糖体(以及它们一起的细菌)而不会影响宿主人类的治疗方法。

细菌对药物的耐药性可以通过两种方式出现
1. 通过积累和收集大量基因,并将它们分类以对抗单个细胞中的药物,或
2. 当越来越多的基因被组织在“多药外排泵”下时,(Hiroshi,2009,第 119 页),并且正在迫使大量药物排出。


尽管有 100 多种抗生素,但大多数抗生素仅来自少数几种药物。这些是主要类型的抗生素
1. 氨基糖苷类它们主要用于对抗由革兰氏阴性细菌引起的感染。据说细菌能够提高对它的抵抗力;但是,细菌能够提高对几乎所有事物的抵抗力。氨基糖苷类可能引起的副作用包括肾脏或耳朵损伤。氨基糖苷类主要的治疗结果是它阻止了细菌在你的身体里产生蛋白质。
2. 头孢菌素 头孢菌素有三种不同的生成类型。代数越高,其对革兰氏阴性菌的抗菌作用就越大。特别是晚代头孢菌素,细菌更不容易对其产生耐药性。这显然是一个优势,因为它意味着药物在更长的时间内会更有效。
3. 氟喹诺酮 这种类型的抗生素对几种不同的细菌有效,但它主要用于治疗尿路感染(UTI)、呼吸道感染和皮肤感染。使用氟喹诺酮时,它会导致恶心、腹泻、呕吐或胃部轻微疼痛。它的作用机制是抑制细菌合成 DNA 的能力,从而使细菌更难繁殖。
4. 青霉素 这是 1929 年亚历山大·弗莱明发现的第一种抗生素。青霉素主要用于治疗牙周感染、呼吸道感染、淋病、尿路感染(UTI)和皮肤感染。青霉素的作用机制是阻碍细菌细胞壁的生长。最终,细胞壁会破裂,由于这个原因,细菌会在一段时间内被杀死。虽然你可能会对这种抗生素产生过敏反应,但医生认为青霉素非常安全。
5. 四环素 当它们在 1940 年代首次被发现时,四环素是最常见的抗生素。四环素最常见的用途是治疗上呼吸道感染、性病、莱姆病、轻度痤疮和伤寒。它们在大多数情况下用于对抗多种不同的细菌感染。多西环素和米诺环素都是四环素。
6. 大环内酯类 这类抗生素主要用于治疗胃肠道和其他部位的感染。它们与易感细菌的核糖体结合,这将有助于避免任何蛋白质的产生。这有助于随着时间的推移杀死所有有害细菌。大环内酯类唯一的实际副作用是它们会导致胃部不适,但没有严重的副作用。
杆菌肽是由细菌微生物产生的抗生素。杆菌肽抗生素是从枯草芽孢杆菌中分离出来的。杆菌肽是一种窄谱肽类抗生素,它能抑制革兰氏阳性菌的细胞壁合成。杆菌肽是用于治疗由金黄色葡萄球菌和链球菌属细菌引起的皮肤感染的百多邦软膏的主要成分。这种抗生素最初是在 1945 年分离出来的。它用于局部使用,涂抹在皮肤上,用于消毒轻微伤口,不能口服。杆菌肽通常不口服,因为它作为局部抗生素更有效。 [3]
杆菌肽对革兰氏阳性菌最有效,因为革兰氏阴性菌在细胞壁中有一层外膜。杆菌肽通过靶向细菌前体磷脂,一种存在于细胞质膜中的脂质,通常用于将肽聚糖的二糖单元跨细胞膜转运,以添加到正在生长的肽聚糖链中,从而杀死细菌。 [1]
在细菌肽聚糖的形成中,NAM 和 NAG 糖在细胞内以新磷酸化的细菌前体磷脂的形式连接在一起。然后 NAM-NAG 二糖单元通过细菌前体磷脂转运到细胞外膜。在二糖单元添加到正在生长的肽聚糖链后,细菌前体磷脂从 NAM-NAG 单元中去除。然后该细菌前体磷脂释放一个磷酸,然后去磷酸化的细菌前体磷脂向细胞膜的胞质部分移动,重复该循环。
杆菌肽通过在二糖单元添加到正在生长的肽聚糖后立即与细菌前体磷脂结合而起作用。这抑制了细菌前体磷脂的去磷酸化,从而阻止细菌前体磷脂在细胞内接受 UDP-NAM。这样,杆菌肽导致细菌肽聚糖细胞壁的合成停止。正在生长的细菌将由于细胞内部压力增加而裂解。 [1]
副作用几乎没有,但极少数情况下可能会发生过敏反应、皮疹、瘙痒、头晕或呼吸困难。
利福霉素 B 是在 1957 年与其他六种利福霉素(A、C、D、E、S 和 SV)一起发现的。利福霉素 B 是第一个商业化使用的利福霉素。它在 1960 年代被誉为解决耐药性结核病的解决方案。
利福霉素 B 选择性地与细菌 RNA 聚合酶结合,以抑制 mRNA 的翻译。它与聚合酶的β亚基结合,该亚基位于 Mg2+ 活化位点附近。当这种情况发生时,正在形成的肽链无法从 RNA 聚合酶中出来,而是被卡住了。但是,需要注意的是,利福霉素 B 只有在与刚刚开始翻译的 RNA 聚合酶结合时才有效,否则它无法阻止已经生长的肽链。
利福霉素 B 在治疗由分枝杆菌引起的细菌感染方面非常有效,分枝杆菌具有非常厚而蜡质的细胞壁,这使得靶向细胞壁的抗生素(青霉素和万古霉素)无效。这些疾病包括结核病和麻风病。
放线菌素 D 是第一个被发现具有抗癌特性的抗生素。它首次使用是在 1964 年。它被用作化疗药物。
放线菌素 D 的结构使其能够模拟核碱基,并将其插入鸟嘌呤和胞嘧啶之间。这阻止了 RNA 聚合酶继续转录过程中的延伸过程。然而,问题是放线菌素 D 不仅与细菌 DNA 结合,还与任何 DNA 非选择性结合。
放线菌素 D 被用作化疗药物,以抑制 DNA 合成,从而抑制细胞分裂。这使其对不受控制地分裂的肿瘤细胞特别有效。
由于放线菌素 D 并非用于治疗感染,并且它不 specifically靶向病原体 DNA,因此它会抑制正常细胞中的 DNA 合成。其他副作用包括骨髓抑制、脱发、疲劳、口腔溃疡、食欲不振和腹泻。
链霉素是一种氨基糖苷类抗生素,它会影响细菌的翻译。 [1]
链霉素是一种氨基糖苷类抗生素,由某些细菌产生,以保护自身免受竞争细菌的侵害。链霉素之所以特别有效,是因为它具有特异性:它攻击细菌核糖体,破坏细菌的蛋白质合成,但不会攻击包括人体核糖体在内的许多其他生物体的核糖体。因此,链霉素成为了一种完美的抗生素药物,可以控制细菌感染,对我们自身的细胞几乎没有副作用。
机制
[edit | edit source]链霉素与 30S 核糖体亚基的 16S rRNA 和蛋白质 S12 结合,从而抑制 70S 细菌核糖体的形成。在低浓度的链霉素中,翻译可以进行,但核糖体的 A 位点允许不精确的密码子和反密码子的匹配,导致蛋白质的错误翻译。 [1]
细菌耐药性
[edit | edit source]细菌可以通过在 S12 中诱发突变来对抗链霉素,使链霉素无法结合。细菌还可以改变链霉素,使其无法与特定分子结合。 [1]
效果
[edit | edit source]链霉素会影响细菌的蛋白质合成。它会影响信使 RNA 的读取方式,导致翻译错误,并抑制核糖体沿着 mRNA 链的有序移动。它还会阻止核糖体在完成蛋白质合成后循环利用。
四环素
[edit | edit source]四环素是一种会影响细菌翻译的抗生素。使用四环素或多西环素作为抗生素治疗的一个优势是它具有广谱抗菌作用,这意味着这种抗生素可以对抗多种细菌菌株,例如支原体属,立克次体属的立克次体,它会导致落基山斑疹热,以及衣原体属的沙眼衣原体。 [3]
机制
[edit | edit source]四环素通过阻止 tRNA 与 A 位点结合,从而阻止蛋白质的合成。它通过与 16S rRNA 位点结合来实现这一点,这样新的 tRNA 就无法添加到该位点。 [1]
细菌耐药性
[edit | edit source]细菌通过启动一个细菌转运系统将抗生素转移到细菌细胞外部来对抗四环素,这样四环素就无法再影响细胞了。 [1]
氯霉素
[edit | edit source]氯霉素是一种会影响细菌翻译的抗生素。
机制
[edit | edit source]氯霉素通过攻击 50S 核糖体亚基来阻止翻译。在攻击 50S 亚基时,氯霉素会与 23S rRNA 上的肽酰转移酶位点结合。这种肽酰转移酶用于将新的氨基酸与已翻译的蛋白质链结合。因此,氯霉素的结合会阻止肽键的形成。 [1]
细菌耐药性
[edit | edit source]细菌通过产生氯霉素乙酰转移酶来对抗氯霉素,这种酶通过改变抗生素的特性来阻止氯霉素的任何活性。 [1]
红霉素
[edit | edit source]红霉素是一种属于大环内酯类的抗生素,其特征是含有 12-22 个碳原子的内酯环。红霉素或阿奇霉素可以治疗革兰氏阳性菌菌株,例如会导致白喉的棒状杆菌,以及革兰氏阴性菌,例如会导致百日咳的百日咳杆菌。 [3]
机制
[edit | edit source]红霉素通过与肽酰转移酶腔蛋白 L15 和 23S rRNA 相连,附着在 50S 核糖体亚基上。这种结合会导致肽酰-tRNA 从 P 位点弹出。 [1]
细菌耐药性
[edit | edit source]一些细菌通过在 L15 中发生突变来对抗红霉素,使其不再结合。细菌还可以通过使用一种酶使 23S rRNA 区域甲基化来减少红霉素的结合,从而抑制 L15 的结合。 [1]
万古霉素
[edit | edit source]万古霉素于 1952 年从婆罗洲土壤样本中分离出来的东方链霉菌中分离出来,发现它对大多数革兰氏阳性生物体都有效。它在发现之初特别珍贵,因为它对当时许多对其他抗生素有耐药性的生物体有效。它于 1958 年获得美国食品药品监督管理局 (FDA) 批准,并在那个十年里流行起来。由于对毒性的担忧,万古霉素在随后的几年里不再流行。它在 1980 年代重新流行起来,到 1987 年,欧洲和美国开始出现耐药性报告。 [4]
机制
[edit | edit source]这种抗生素通过靶向生长细菌细胞壁上肽聚糖链的末端 D-丙氨酰-D-丙氨酸起作用;它与该丙氨酸二聚体连接,阻止保护性细胞壁的形成。 [5][4]
耐药细菌已经将这个双丙氨酸链修改为D-丙氨酰-D-乳酸或D-丙氨酰-D-丝氨酸(取决于耐药菌种的分类);对这些细菌,万古霉素无效。实验表明,对于某些万古霉素耐药菌株,替考拉宁和庆大霉素的联合用药比单一使用万古霉素或替考拉宁更有效。另一种万古霉素耐药菌株对高剂量的糖肽类药物有反应。最终,一项研究建议使用庆大霉素的联合药物治疗。[4]
没有报道标准的临床用量,但过去常用图表。临床使用万古霉素一直存在的一个问题是,血清浓度没有得到一致的监测或广泛研究。有些医生从不监测血清,偶尔监测,或在患者被开具万古霉素处方时坚持频繁监测。为了有效地使用万古霉素,这是一个需要在未来解决的问题。[4]
青霉素G是一种由真菌微生物产生的抗生素。这种抗生素是从青霉菌属中分离出来的,通过阻止酶产生肽聚糖中的氨基酸交联来抑制真菌的细胞壁合成。因此,这种抗生素用于革兰氏阳性细菌,它们含有厚厚的肽聚糖层。[3]
1. 噻唑烷酮环
2. β-内酰胺环
3. R基团(侧链不同)
yeesh
头孢菌素C是一种由真菌微生物产生的抗生素。这种抗生素用于革兰氏阳性细菌,是从头孢菌属中分离出来的,它是一种霉菌。头孢菌素C可以抑制真菌的细胞壁合成,并含有β-内酰胺基团和用于修饰的R基团。[3]
1. 比青霉素更广谱
2. 比青霉素过敏反应更少
3. 耐青霉素酶(细菌产生的破坏青霉素的酶)
1. 从肠道中吸收不良
2. 药物通过注射进入静脉(静脉注射)或肌肉(肌内注射)进行肠外给药(非口服)
[6]银作为抗菌剂,自19世纪以来就为人所知并使用,1998年的一项研究检验并试图优化这种治疗方法。尽管细菌对其他形式的抗生素会产生耐药性,但还没有报道细菌对抗菌银产生耐药性(尽管重金属耐药性基因确实存在,并且与抗生素耐药性基因相关)。
银离子对细菌有毒,因为它们会阻断细菌功能的几个必要过程:银离子会阻止呼吸酶正常工作,阻断电子传递链的某些部分,并损害一些DNA功能。
过去,乳膏形式的银离子曾被测试,但由于乳膏的性质(它们往往会干涸,患者会发现更换敷料不舒服),效果不理想。非常强的离子溶液被证明会刺激皮肤。在一项由 Wright 等人进行的研究中,对三种不同的银化合物进行测试,这些化合物被送入敷料中:敷料中的硝酸银、敷料中的磺胺嘧啶银和敷料中的纳米银。这些敷料被接种了对几种抗生素都有耐药性的细菌菌株;细菌被分离并重新培养,并测量细菌的重新生长。纳米银敷料最明显地减少了重新生长。它不像另外两种处理方法那样刺激皮肤。此外,血清蛋白(在本研究中通过小牛血清溶液测试)并没有降低纳米银处理的功效。这种处理方法在临床环境中似乎非常有价值。

许多细菌已经对现有的抗生素产生了耐药性。此外,金黄色葡萄球菌(MRSA)等一些疾病对大多数药物无反应,因为它们对大多数抗生素产生了耐药性。这些疾病只能用“储备抗生素”治疗,通常用于威胁生命的全身感染,例如万古霉素。

抗生素是一种抑制微生物生长或破坏微生物的药物。与真核生物的 80S 核糖体相比,细菌有 70S 核糖体,它们利用抗生素来阻断蛋白质的合成。当抗生素能够与细菌中的“蛋白质合成起始复合物”结合并干扰其形成时,这种抗生素是有价值的。我们需要抗生素来对抗细菌。因此,抗生素的存在是为了打断细菌的生长。线粒体中的核糖体和细菌中的核糖体之间的区别是,抗生素会影响其功能。
Nikaido, Hiroshi. "细菌的多重耐药性." Annu. Rev. Biochem. (2009): 119-46. 网络。
图片:维基百科
http://www.emedexpert.com/classes/antibiotics.shtml
http://www.emedicinehealth.com/antibiotics/article_em.htm
http://www.medicinenet.com/bacitracin-topical/article.htm
Davis, Alison. (2006). 细胞内部. 国家卫生研究院,9。
- ↑ a b c d e f g h i j k l Slonczewski, Joan L. Foster, John W. 微生物学:不断发展的科学,第二版,W.W. Norton & Company。2009 年。
- ↑ 美国卫生与公众服务部。细胞内部。2005 年 9 月。<http://www.nigms.nih.gov>。
- ↑ a b c d e Tortora, Gerard J.,Berdell R. Funke 和 Christine L. Case。微生物学:导论。第 10 版。美国加利福尼亚州旧金山:Pearson Benjamin Cummings,2010 年。印刷版。
- ↑ a b c d Levine, Donald P. "万古霉素:历史。" 临床传染病 42 (2006): S5-12。
- ↑ Alison Davis,博士。"健康化学"。2006 年 8 月。美国国立卫生研究院,国立普通医学科学研究所主页。NIH。2012 年 12 月 <http://www.nigms.nih.gov >。
- ↑ J. Barry Wright,博士,Bsc Kan Lam 和博士 Robert E. Burrell。"在细菌抗生素耐药性不断增加的时代进行伤口管理:局部银治疗的作用。" 美国感染控制杂志 26.6 (1998): 572-577。
前药是一类新型药物,在给药后不具有活性。吸收后,药物被激活。这一过程称为生物活化,是由体内代谢发生的。前药的用途在于它们可以用于避免与标准药物吸收相关的负生理效应。[1] 这一类药物的潜在益处是巨大的。它们可以改善药物靶向性和在需要的地方提高活性成分的浓度。可以通过使用前药克服的标准药物的问题包括:口服吸收不足、溶解度有限以及与给药相关的刺激。[2]
不良性质
[edit | edit source]- 物理性质:水溶性差、亲脂性低和化学不稳定 - 药代动力学性质:跨生物膜分布差,是首过代谢的良好底物,在需要长期效果时快速吸收/排泄。
前药分类
[edit | edit source]从人体如何将前药转化为最终的活性药物形式来看,实际上存在两种主要类型的前药。
一种类型的前药称为 I 型前药。这种类型的前药在细胞内被生物活化。降脂他汀类药物和抗病毒核苷类似物都是 I 型前药的例子。还有一种前药称为 II 型前药。与 I 型前药不同的是,II 型前药在细胞外被生物活化,特别是在人体的循环系统或消化液中。例如,抗体或病毒定向酶都是 II 型前药。它们也常用于免疫疗法或化疗。
前药亚型
[edit | edit source]还可以看到,I 型和 II 型前药可以被归类为亚型。例如,I 型具有细胞内生物活化位点,并且包含 I 型 A 和 I 型 B 亚型。I 型 A 通常位于治疗靶组织或细胞中。这方面的例子可以是己烯雌酚二磷酸酯或 6-巯基嘌呤。然后是 I 型 B,它位于肝脏、胃肠道粘膜细胞或肺部的代谢组织中,这种亚型的例子可以是海洛因、苯妥英钠或卡托普利。
此外,就像 I 型可以被分类为不同的亚型一样,II 型前药也做同样的事情。例如,II 型具有细胞外生物活化位点,并且包含 II 型 A、II 型 B 和 II 型 C 亚型。II 型 A 通常存在于胃肠道液中,这方面的例子可以是柳氮磺胺吡啶。II 型 B 存在于全身循环和其他细胞外液室中。氯霉素琥珀酸酯或滴眼液都是 II 型 B 前药的例子。最后但并非最不重要的是,II 型 C,如 ADEPT、VDEP 或 GDEP,都存在于治疗靶组织或细胞中。
因此,存在不同类型和种类的前药,因为一种类型的前药包含几个或多个亚型,这些亚型通常存在于人体的不同位置。
前药设计步骤
[edit | edit source]- 确定药物递送问题,并确定所需的理化性质 - 选择将使前药获得所需运输性质并易于在所需生物区室中裂解的转运部分
设计与结构
[edit | edit source]目前,许多前药主要利用羟基、胺基和羧基。酯类在商业化前药中最为常见,例如奥司他韦。也有人研究将更不常见的基团用于前药,例如硫醇和亚胺。[3] 前药生物转化为其活性母体药物是通过水解酶的酶活性实现的。前药可以根据催化反应的酶的特定特性来设计生物转化,特别是底物识别。[4]
示例
[edit | edit source]前药伐昔洛韦最近被开发出来,试图阻止三种不同类型的单纯疱疹病毒的传播:1 型、2 型和水痘带状疱疹病毒。这种药物含有两个酯基,需要两种酶才能将该分子生物转化为其活性形式,这一过程主要在肝脏中进行。
替诺福韦酯(替诺福韦二吡伏酯),另一种二酯类前药,负责抑制针对乙型肝炎病毒的核苷逆转录酶。这种药物的吸收发生在口服后,而且速度非常快,在短短 3/4 小时后达到最大吸收。
维瑞德是一种基于碳酸盐的前药,与前面提到的基于酯的前药不同。它的目的是抑制 HIV 病毒和乙型肝炎病毒中的核苷转录酶。这种前药中所含的碳酸盐基团被发现比前面提到的前药中所含的酯基更稳定,而最大吸收速度较慢。
参考文献
[edit | edit source]- ↑ http://www.chem.memphis.edu/parrill/chem4315/2005/Prodrugs.pdf
- ↑ http://www.nature.com/nrd/journal/v7/n3/full/nrd2468.html
- ↑ http://epublications.uef.fi/pub/urn_isbn_978-951-27-0634-1/urn_isbn_978-951-27-0634-1.pdf
- ↑ Imai, Teruko 和 Masakiyo Hosokawa。"利用羧基酯酶活性进行前药设计:羧基酯酶在哺乳动物组织中的催化性质和基因调控。" 农药科学杂志 35.3 (2010): 229-239。
http://en.wikipedia.org/wiki/Prodrug
概述
[edit | edit source]选择性5-羟色胺再摄取抑制剂(SSRI)是一类抗抑郁药物,其作用机制是通过抑制5-羟色胺重新摄取到突触前神经元,从而增加突触间隙中5-羟色胺的水平。它们对其他单胺类转运蛋白(如去甲肾上腺素和多巴胺)具有不同的亲和力,这造成了它们名称中的“选择性”一词。SSRI 已成为最流行的抗抑郁药物,取代了具有更多副作用和略微降低疗效的单胺氧化酶抑制剂。然而,关于 SSRI 的实际益处仍然存在争议,因为一些荟萃分析表明,与安慰剂相比,抗抑郁药物的效果在统计学上显著,但在临床上并不显著。 [1] SSRI 是最常见的抗抑郁药物,因为它可以治疗中度到重度抑郁症,风险相对较低,副作用较少。选择性5-羟色胺再摄取抑制剂可以制成缓释 (XR) 或控释 (CR) 型。这些类型药物的优势在于它们可以缓慢地长时间释放药物。因此,与药物相关的起伏更少,可以平稳地过渡到药物治疗。这些药物可以用于除抗抑郁剂之外的其他目的。SSRI 对每个人都有不同的影响,因为每个人的大脑对药物的化学成分都有不同的反应。可能的副作用包括口干、恶心、紧张、头痛、腹泻、嗜睡、失眠等。 [2]
药理动力学
[edit | edit source]SSRI 抑制5-羟色胺重新摄取到突触前神经元,从而增加突触间隙中可利用的5-羟色胺。然而,抗抑郁作用需要连续服用 SSRI 数周才能显现。原因是5-羟色胺水平升高会产生两种神经变化:首先,它会导致 5-HT2A 受体下调,从而改变神经递质与受体之间的比例。其次,它还会触发突触前间隙上的自身受体,导致细胞因对可利用的 5-羟色胺水平升高做出反应而下调而产生更少的 5-羟色胺。或者,SSRI 还会影响其他途径,例如突触后细胞上的环状 AMP (cAMP),导致脑源性神经营养因子 (BDNF) 的释放。这种蛋白质分泌以调节神经网络以及突触可塑性。 [3]
一些 SSRI 的例子
[edit | edit source]- 西酞普兰 (Celexa, Cipramil, Cipram, Dalsan, Recital, Emocal, Sepram, Seropram, Citox, Cital)
- 达泊西汀 (Priligy)
- 艾司西酞普兰 (Lexapro, Cipralex, Seroplex, Esertia)
- 氟西汀 (Prozac, Fontex, Seromex, Seronil, Sarafem, Ladose, Motivest, Flutop, Fluctin (EUR), Fluox (NZ), Depress (UZB), Lovan (AUS), Prodep (IND))
- 氟伏沙明 (Luvox, Fevarin, Faverin, Dumyrox, Favoxil, Movox)
- 依达普林 (Upstene) (已停产)
- 帕罗西汀 (Paxil, Seroxat, Sereupin, Aropax, Deroxat, Divarius, Rexetin, Xetanor, Paroxat, Loxamine, Deparoc)
- 舍曲林 (Zoloft, Lustral, Serlain, Asentra)
- 维拉佐酮 (Viibryd)
- 齐美替丁 (Zelmid, Normud) (已停产)
- ↑ 严重和焦虑性抑郁症:结合临床亚型定义以确定对选择性5-羟色胺再摄取抑制剂反应不同的患者1
- ↑ SSRI,2012 年 10 月 28 日
- ↑ 选择性5-羟色胺再摄取抑制剂途径2
大多数人,如果不是所有人,都服用过药物。他们服用药物来缓解疼痛、疾病或酸痛。通过大量的研究,科学家们能够确定药物在人体内经过的四个基本阶段。这四个阶段是吸收、分布、代谢和排泄。整个过程有时缩写为 ADME。
吸收
[edit | edit source]药物通过人体的第一步是吸收。药物进入人体的途径很少。最常见的方法是“口服”,无论是片剂(药丸)还是液体(用勺子、杯子或注射器)。在家里,许多人使用这种方法。另一种方法是“肌肉注射”,这意味着将药物注射到手臂肌肉中。例如,当流感流行时,每个人通常都会通过这种方法注射流感疫苗。药物进入人体的另一种方法是“皮下注射”,即注射到皮肤下。另一种途径是“静脉注射”,这通常发生在接受化疗的人身上。药物通过身体的静脉进入。药物进入人体的最后一种方法是“透皮”,这实际上不是插入到人体中。这种方法的例子是佩戴皮肤贴剂。药物通过皮肤吸收并通过我们的皮肤毛孔进入人体。
在药物通过人体的所有步骤中,吸收是最难克服的障碍,因为每个人都是不同的。如果身体排斥药物,一些药物可能无法进入人体。如果药物要遍布全身,它需要首先被吸收。患者的身体必须接受药物才能发挥作用。药物给药可能会出现并发症,因为药物可能不会通过身体的正确途径。
分布
[edit | edit source]药物被吸收后,药物通过人体的下一步是分布。药物从身体的一侧进入,但目的是治疗身体的另一部分,这可能是身体的中间部分(或很难直接注射的地方)。血液是药物从进入药物的表面到目的地的运输工具。药物必须克服的一个障碍是副作用。如上所述,我们每个人都是不同的。一种对一个人有效的药物可能对另一个人有效,也可能无效。人们对药物的反应可能不同,会出现一种或多种副作用。有些人对某些药物过敏。许多因素可能会阻止药物在体内分布。“血液中蛋白质和脂肪分子的存在”会攻击药物,阻止药物发挥作用。药物还有其他并发症,尤其是如果它们旨在进入“中枢神经系统 (CNS)”,即大脑和脊髓所在的地方。中枢神经系统有一个称为“血脑屏障”的障碍,保护中枢神经系统免受危险药物的侵害。药理学家已经找到将物质插入血脑屏障的方法。
代谢
[edit | edit source]药物到达目的地后,必须“代谢”。药物通常在肝脏中代谢。肝脏被选中胜过任何其他器官,因为它是一个“连续和疯狂,但仔细控制的活动场所”。所有进入血流的物质都会直接进入肝脏,并在该位置,药物被改变以适应身体中的适当功能。“生物转化”是肝脏中发生的改变的产物。它们也被称为酶。酶是某些蛋白质,具有特定的能力,当与底物结合时,它可以执行其功能。
排泄
[edit | edit source]酶完成其功能后,分解产物的化学活性低于原始分子。这些产物被称为代谢物。肝脏被称为“解毒”器官,因为它分解酶。有时药物代谢物本身可能具有化学活性,这种活性可能取代或可能不取代原始药物的活性。当酶完成其功能后,它现在可以通过排泄进行,排泄通常通过肠道进行。
参考
[edit | edit source]参考文献:戴维斯,艾莉森。(2006)。药物设计。美国国立卫生研究院,11-13。
他汀类药物
[edit | edit source]
概述
[edit | edit source]他汀类药物,也被称为 3-羟基-3-甲基戊二酰辅酶 A (HMG-CoA) 还原酶 (HMGCR) 抑制剂,是一类药物,通过减少肝脏胆固醇的产生来降低血液中的胆固醇水平。他汀类药物是抑制 HMGCR 酶的抑制剂,HMGCR 酶负责胆固醇的产生。由于高胆固醇与心脏病风险有关,因此胆固醇水平升高的人可能会发现他汀类药物对预防有益。最畅销的他汀类药物是阿托伐他汀,商品名为立普妥。立普妥由辉瑞公司生产,在稳定斑块和预防中风方面非常有用。
历史
[edit | edit source]1971年,远藤章,开始研究寻找抑制 HMGCR 的抑制剂,以降低肝脏中的胆固醇水平。他和他的团队认为,可能存在微生物产生这种酶抑制剂来防御其他生物。第一个分离出来的物质是美伐他汀,一种由真菌“青霉属的柠檬青霉菌”产生的分子。到1976年,另一种他汀类药物,被称为洛伐他汀,从真菌“曲霉属的土曲霉菌”中分离出来。几年后,其他分离出来的他汀类药物开始进入市场,包括辉瑞公司在1985年推出的立普妥。
之后,胆固醇与心血管疾病之间的联系,被称为脂类假说,已经被提出。胆固醇是一种由肝脏产生的蜡状物质,也存在于某些食物中,它是制造维生素D和一些激素、产生帮助人们消化脂肪的胆汁盐以及构建细胞壁所必需的。然而,胆固醇过高并不利,因为这会导致心脏病发作。在 1984 年的冠心病初级预防试验中,胆固醇研究员丹尼尔·斯坦伯格指出,低脂饮食和难以忍受的药物,如考来烯胺、氯贝特和烟酸,都是治疗方面的各种饮食措施。根据斯坦伯格的说法,任何能降低胆固醇的东西都能大大降低患心脏病和心绞痛等严重问题的风险。然而,包括心脏病专家在内的许多医生仍然对斯坦伯格的理论不相信。
为了推广他汀类药物的使用,默克公司向公众和医生宣传了高胆固醇水平的危害,并说明他汀类药物是安全的,并且可以延长寿命。很快,人们开始熟悉自己的胆固醇数值,并了解了什么是好胆固醇和坏胆固醇之间的区别。随着这种情况的发生,竞争对手的制药公司开始生产自己的他汀类药物,例如由三共制药和百时美施贵宝公司生产的普伐他汀(Pravachol)。在 1994 年,默克公司赞助的一项研究,即“斯堪的纳维亚辛伐他汀生存研究”(也称为“4S”)的结果公布。之后,默克公司开始销售辛伐他汀(Zocor),该药物被 4,444 名患有心脏病和高胆固醇的患者使用。仅仅五年,研究得出结论,许多患者的胆固醇水平降低了 35%,他们死于心脏病的可能性降低了 42%。1955 年,默克公司凭借 Zocor 和 Mevacor 赚取了超过十亿美元的利润。2006 年,远藤章获得了日本奖,2008 年获得了拉斯克-德贝基临床医学研究奖。
谁应该服用它?
他汀类药物用于预防和治疗动脉粥样硬化,这会导致胸痛、心脏病发作、中风和相关死亡。因此,胆固醇水平异常升高或有心脏病史的人应该考虑服用他汀类药物。但是,在开始他汀类药物治疗之前,锻炼和改变饮食可能是降低胆固醇的第一步策略。
他汀类药物的例子
- 阿托伐他汀(立普妥,Torvast)
- 氟伐他汀(Lescol,Lescol XL)
- 普伐他汀(Pravachol,Selektine,Lipostat)
- 瑞舒伐他汀(Crestor)
- 辛伐他汀(Zocor,Lipex)
- 匹伐他汀(Livalo,Pitava)
- 西立伐他汀(Lipobay,Baycol)
- 洛伐他汀(Mevacor,Altocor,Altoprev)
- 美伐他汀(康柏汀)
- 辛伐他汀+依折麦布(Vytorin)
- 洛伐他汀+烟酸(Advicor)
- 辛伐他汀+烟酸(Simcor)
- 阿托伐他汀+氨氯地平(Caduet)
机制
所有他汀类药物的作用机制都是通过抑制 HMGCR,而 HMGCR 催化胆固醇合成过程中 HMG-CoA 到甲羟戊酸的转化。不同的他汀类药物在降低胆固醇方面的效力不同(阿托伐他汀(立普妥)和瑞舒伐他汀(Crestor)是最有效力的他汀类药物,而氟伐他汀(Lescol)是目前市场上效力最弱的他汀类药物)。

副作用
常见的副作用包括:头痛、恶心、呕吐、便秘、皮疹、腹泻、肌肉无力和肌肉疼痛。在极少数情况下,会发生严重的,甚至可能是致命的副作用,例如肝衰竭和横纹肌溶解症(肌肉退化)。这些副作用可能与其他药物的相互作用有关,这些药物会增加他汀类药物的生物利用度。众所周知,他汀类药物会增加患 2 型糖尿病的风险,并且进一步讨论了它对认知功能的影响。一方面,他汀类药物可能会导致工作记忆受损,另一方面,他汀类药物可能会降低患阿尔茨海默病等痴呆症的风险。
参考文献
- http://upload.wikimedia.org/wikipedia/commons/9/99/Simvastatin.png
- http://www.news-medical.net/health/Statin-History.aspx
- http://www.medicinenet.com/statins/article.htm
- http://en.wikipedia.org/wiki/Statin
- http://upload.wikimedia.org/wikipedia/commons/2/25/Cholesterol.png
- http://kidshealth.org/teen/food_fitness/nutrition/cholesterol.html


咖啡因是最流行的药物,被用来维持一定的精神稳定,比如保持清醒、改变大脑功能和情绪。它存在于我们日常饮用的许多饮料中。包括但不限于苏打水、咖啡和茶。许多人甚至没有意识到自己每天摄入多少这种药物,而且大多数人并不认为它是一种危险或值得怀疑的东西。虽然咖啡因被轻视,但我们许多饮料中含有大量这种物质。例如,根据安东尼·温斯顿的文章“咖啡因的神经精神效应”,它表明一杯普通咖啡中含有 100 毫克,速溶咖啡中含有 75 毫克,而我们每天喝的茶中含有 50 毫克(温斯顿,第 432 页)。
通常情况下,咖啡因在摄入后一小时内开始影响大脑和身体,并在 3 到 4 小时后开始消退。
咖啡因主要用于提高和增强精神功能,但过度使用会导致一种叫做咖啡因中毒的有害状态。根据温斯顿的说法,“咖啡因中毒的特点是烦躁不安、激动、兴奋、思维混乱、言语混乱和失眠”(第 1 页)。在这些症状之后,可能会出现许多其他疾病,例如焦虑、睡眠障碍、饮食失调等等。
咖啡因(1, 3, 7-三甲基黄嘌呤)是一种存在于植物中的天然产物。虽然植物种子和叶片中咖啡因的实际含量在不同物种之间差异很大,但咖啡因被认为是最丰富的天然存在的嘌呤生物碱,这意味着它源自一种或多种嘌呤核苷酸。生物碱是一大类化合物,主要存在于植物中,含有碱性氮原子。由于其碱性性质,这些化合物通常以盐的形式存在。主要咖啡因来源包括咖啡植物的种子(阿拉伯咖啡),茶叶(茶树)和可乐果。人们普遍认为,咖啡因被人类食用后是一种兴奋剂,其主要作用集中在中枢神经系统。[1] 植物利用咖啡因作为防御手段。它对植物起着杀虫剂的作用,因为它可以阻碍代谢途径,从而阻止了许多潜在的威胁。[2]
咖啡因的影响:如何以及为什么?
摄入咖啡因后,身体会感到清醒和精神焕发。咖啡因对身体产生影响的原因是它的结构与腺苷和环腺苷单磷酸 (cAMP) 类似。由于结构相似,咖啡因可以潜在地与通常与腺苷及其衍生物结合的受体结合。在体内,腺苷在调节大脑及其活动方面发挥作用。当大脑中积累了大量的腺苷时,它们开始与大脑受体结合,从而引起导致嗜睡和昏昏欲睡的反应。然而,当咖啡因进入人体时,由于其结构相似,它会与相同的受体结合,这阻止腺苷与这些受体结合,从而延缓身体感到困倦。

cAMP 是一种第二信使,负责体内涉及血压和氧气的过程。它会增加输送到大脑的氧气量以及血压,这两者都会使身体保持清醒。然而,体内存在一种酶,即环核苷酸磷酸二酯酶 (cAMP-PDE),它会分解 cAMP 并消除它对身体的抗嗜睡作用。由于其与 cAMP 结构的相似性,咖啡因可以阻止 cAMP-PDE 分解 cAMP,并使抗嗜睡作用持续更长时间。

疾病

- 睡眠障碍
失眠有时是过量咖啡因的结果。在临睡前服用咖啡因可能会延长清醒时间,导致难以入睡。此外,根据温斯顿的说法,“它会减少睡眠周期的早期阶段的慢波睡眠,并可能减少睡眠周期的后期阶段的快速眼动睡眠”(第 2 页)。总之,如果随意服用咖啡因,清醒是其引起的睡眠障碍的症状。
- 饮食失调
咖啡因也是厌食症和神经性贪食症的原因。研究表明,患有这些饮食失调的人经常大量摄入咖啡因。这种药物会导致食欲下降和新陈代谢加快。此外,它也可能对心脏有害,并增加患骨质疏松症的风险。
合成
在这个合成过程中,“SAM”指的是 S-腺苷-L-蛋氨酸,它会导致甲基基团的添加。[2]

咖啡与体重减轻的联系:咖啡因是一种兴奋剂,它会刺激中枢神经系统,在处理疲劳时有所帮助,有助于防止嗜睡和疲倦,并改善警觉性、情绪和新陈代谢。重要的是要监控咖啡因摄入量,并保持均衡的饮食。咖啡是咖啡因的常见来源。每天喝两到四杯咖啡被认为是健康的。能量饮料和含咖啡因的碳酸饮料含有危险量的咖啡因。每天喝超过两杯就足以发挥其预期作用。喝得更多可能会导致严重问题。咖啡因也与体重减轻有关。咖啡因可能有助于减掉几磅,或阻止进一步的体重增加。咖啡因是人们依赖于减肥的危险减肥药中的成分之一。人们认为咖啡因有助于减肥,因为它可以:抑制食欲、提高新陈代谢和燃烧卡路里的机会,以及诱导水分流失。咖啡因可能在短时间内抑制食欲,但它似乎与长期抑制食欲没有任何关联。咖啡因可能会实施和诱导产热作用,这是身体在消化食物时产生热量的过程。咖啡因也可能会让身体保持警觉,并产生行为变化,这可能有助于运动并加速卡路里燃烧。咖啡因燃烧脂肪,这会防止替代品(糖原、葡萄糖、氨基酸)被燃烧。这会使血液中的糖分保持稳定,并使身体能够在不感到饥饿的情况下持续更长时间。咖啡因也是运动前流行的摄入选择。如果有人在运动前摄入咖啡因,研究表明,咖啡因会隔离额外流动的脂肪,并针对脂肪并将其作为能量来源,而不是碳水化合物或现有的肌肉。咖啡因会针对脂肪并将其分解,并立即快速燃烧。但是,在运动期间要喝大量的水以防止脱水。特别是咖啡,可能会导致饮食失调,例如厌食症和贪食症,因为人们认为咖啡没有卡路里和碳水化合物。咖啡实际上含有 0.5-0.9 克碳水化合物。咖啡苦味也可能有助于一个人减少对食物的渴望和对食物的需求,并说服某人自己并不饿。众所周知,咖啡因还可以阻止脂肪在细胞中堆积。有关于咖啡因抑制酶及其在脂肪合成中的作用的研究。咖啡因是众多减肥药的主要成分。有饮食失调的人会求助于咖啡因和咖啡来获得能量并满足饥饿感。但是,在这种情况下,它弊大于利。咖啡因也可能增加排尿量,因为它是一种利尿剂。排尿量增加可能有助于减轻水分重量和肿胀。但是,通过去除液体来减轻体重,效果只会持续很短时间。咖啡因不适合作为膳食替代品,也不适合作为唯一的减肥方法。依赖咖啡因减肥不是一种健康或有效的方法。由于咖啡因的产热特性是减肥药物采用的特性之一,因此可以选择咖啡因作为产热作用的替代品,而不是减肥药物本身。依赖咖啡因会导致极端的健康影响,包括身体和精神方面。它会增加失眠、紧张的几率,使人感到焦躁不安、不稳定、心率加快和钾缺乏。孕妇应该避免大量摄入咖啡因,因为咖啡因会通过血流直接到达胎儿。重要的是要考虑到,为了实现有效的减肥,饮食和运动是两个最关键的方面和因素。依赖和过量摄入咖啡因会产生破坏性的影响,可能会导致身体增重,而不是预期减轻。
提取
[edit | edit source]茶叶
[edit | edit source]咖啡因可以从茶叶中提取出来并分离出来。由于咖啡因可溶于水和有机溶剂,因此可以首先通过在热水中进行固液萃取从茶叶中提取咖啡因,然后通过用极性非质子有机溶剂(如二氯甲烷)萃取进一步分离。在开始萃取过程之前,应该分析茶叶的成分
- 纤维素:纤维素是一种刚性且不溶性的结构成分,构成了叶片的大部分。这种线性聚合物由 D-吡喃葡萄糖单元组成,这些单元在碳 1 和 4 之间连接。由于其高分子量,纤维素尽管具有羟基,但在水中也不溶解。

纤维素塞塞尔

~在用热水进行固液萃取茶叶时,纤维素很容易去除,因为当咖啡因被萃取到水中时,纤维素成分会留在后面。纤维素保留在茶叶中,而咖啡因和其他水溶性成分被萃取出来。
- 蛋白质和色素:色素和蛋白质都高度溶于水,因此不会对咖啡因的萃取造成问题。在最初的固液萃取中,蛋白质和色素会与咖啡因一起被萃取出来。但由于它们是非极性的,因此在将咖啡因萃取到二氯甲烷(不溶于水)的第二步萃取中,咖啡因会被萃取出来,而不会萃取蛋白质和色素。它们会留在水相中,而咖啡因会被萃取到这种不溶于水的二氯甲烷中。
- 单宁:单宁是多酚类化合物,具有可变的分子量,是茶叶苦味的原因。这些化合物由 D-葡萄糖单元组成,这些单元与没食子酸单元通过酯键连接。单宁会对咖啡因的萃取造成问题,因为它可溶于水和有机溶剂。但是,这个问题可以通过用弱碱(例如碳酸钙)裂解酯键来解决。这会导致单宁分解成葡萄糖和没食子酸的钙盐,它们都不会被萃取到有机溶剂中。
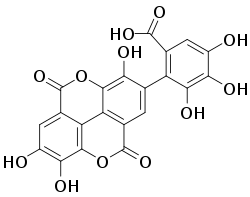
黄没食子酸二内酯 - 皂苷:皂苷是既具有极性又具有非极性特性的化合物。这种二元性使萃取过程复杂化,因为有机化合物在水中变得更易溶,这会导致乳液的形成。乳液使得难以有效地分离溶剂层。乳液可以通过离心或增加水层的极性来去除。例如,添加氯化钠盐会降低有机化合物在水中的溶解度。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg%26page%3D1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
提取过程
[edit | edit source]

首先,可以通过固液萃取从茶叶中提取咖啡因。这可以通过将茶包在水中和弱碱(例如碳酸钙)中煮沸来实现,以破坏单宁中的酯键。然后可以将液体咖啡因转移出来并进行真空过滤。过滤后的咖啡因可以转移到离心管中,并准备用有机溶剂进行第二次萃取。当加入有机溶剂时,应让两层分离,然后再去除所需的有机层。如果形成乳液,则应进行离心并相应地添加盐。液体咖啡因应该用有机溶剂进行多次萃取。收集的有机萃取物可以在热板上用沙浴蒸发,以获得“粗制”产品。由于咖啡因具有升华的特性,因此可以进一步纯化该产品。升华是指化合物从固态直接变为气态,而无需经过液态。任何杂质都不会与咖啡因一起升华。 [3] 杂质不能与咖啡因一起升华,因为它们需要不同的温度和压力。从本质上讲,咖啡因从粗产品中升华出来,使实验者能够收集咖啡因并留下杂质。 [4]

- 升华
升华器可以以多种方式设置,但主要目标是加热升华室内的固体,使其达到气态,并收集到插入升华室的管子或装置中。升华完成后,可以取出管子,并将收集的咖啡因取下。 [3] 该管子连接到真空装置,该装置会降低装置中的总压力。这种压力的降低确保咖啡因升华而不是熔化然后汽化。此外,压力的降低不会使杂质升华,使实验者能够收集纯净的咖啡因晶体。 [4]
MVIE 提取方法
[edit | edit source]微波真空冰水萃取 (MVIE) 是一种利用微波功率提取咖啡因的方法。它已在绿茶叶中进行了测试,并获得了很高的去除率。该过程取决于溶剂与固体的比例、微波功率和萃取时间。如果能正确执行技术并控制时间,它可以提取大约 87.6% 的咖啡因。与热水萃取相比,MVIE 被证明是提取咖啡因的更有效方法。
- 基本步骤
茶叶和水的混合物经过微波萃取,然后进行真空冰水萃取。然后将样品放入真空烘箱中,蒸汽会在一个单独的容器中收集。进行多次萃取以从茶叶中提取足够的咖啡因。
- 因素
微波功率:当功率从 160 瓦增加到 350 瓦时,提取的咖啡因含量分别为 22% 和 46.6%。这表明,增加微波功率会增加提取的咖啡因含量。功率增加会提高内部热量,最终加速溶剂从叶片中释放到溶液中的过程。但是,任何高于 350 瓦的功率都会导致相反的结果——咖啡因提取量减少。
溶剂与固体比例:产生最高提取量的比例为 10:1(溶剂毫升数:固体克数)。
MVIE 提出了一种从茶叶中提取咖啡因的全新有效方法。
Mohrig,Jerry R.,Christina Noring Hammond 和 Paul F. Schatz。《有机化学技术》。纽约:W. H. Freeman and Company,2010 年。印刷版。
P. Nawrot、S. Jordan、J. Eastwood、J. Rotstein、A. Hugenholtz 和 M. Feeley(2003):咖啡因对人体健康的影响,食品添加剂和污染物,20:1, 1-30
Winston,Anthony P. "咖啡因的神经精神效应"。精神治疗进展 11 (2005): 432–39。网页。
Lou Z.、Er C.、Li J.、Wang H.、Zhu S. 和 Sun J. (2011):微波增强真空冰水提取法去除绿茶中的咖啡因,分析化学学报,49-53
图片:维基百科共享资源
- ↑ a b Ashihara,H 和 Ashihara,H. (2004)。植物中咖啡因的分布和生物合成。生物科学前沿,9(1-3), 1864-.
- ↑ a b Mohanpuria,P 和 Mohanpuria,P. (2009)。茶叶 [Camellia sinensis (L.) O. Kuntze] 中的咖啡因生物合成和降解受发育和季节调节。分子生物技术,43(2), 104-.
- ↑ a b Mohrig,Jerry R.,Christina Noring Hammond 和 Paul F. Schatz。《有机化学技术》。纽约:W. H. Freeman and Company,2010 年。印刷版。
- ↑ a b 提取,2012 年 10 月 28 日
麦角酸二乙酰胺,俗称 LSD,已知会导致迷幻幻觉和幻视。它并不具有成瘾性,也不会造成脑损伤。LSD 通常用于娱乐目的,以增强创造力和体验“出体”的感觉。1938 年,阿尔伯特·霍夫曼在制造二乙酰胺的不同衍生物时发现了 LSD。它在 1943 年被意外地发现了迷幻特性。在 20 世纪 50 年代,LSD 被引入医疗界,作为一种工具来制造暂时的精神病状态,并与精神治疗相结合。在 20 世纪 60 年代,公众开始将 LSD 用于娱乐目的,直到今天它仍然是一种主要毒品。[1]。
LSD 阻断大脑中的血清素。血清素是一种神经递质,负责调节情绪、肌肉收缩和其他认知功能。LSD 阻断血清素的原因是它们在结构上的相似性,LSD 被误认为血清素,然后被送往突触间隙而不是血清素。与许多其他迷幻剂一样,LSD 在其分子结构中有一个取代的吲哚环,这导致了它的迷幻作用。许多感知变化会改变认知和视觉感官系统,以及时间感、身体意象和自我意识的变化。记忆也受到很大的影响。一次典型的“迷幻之旅”可以持续 6 到 10 个小时。人类的半衰期约为 175 分钟,但它在身体的不同结构中以不同的速度代谢。自 20 世纪 70 年代以来,没有进行过太多临床测试,部分原因是受试者在服用 LSD 后“变得过于虚弱,无法配合”。[1]
LSD 晶体形式的分子构型已通过 X 射线衍射技术确定。构型显示应力和空间位阻。血清素和 LSD 的结构在一定程度上是相似的,这可以通过两种分子中存在的吲哚环来证明。因此,由于结构的相似性,两种分子具有相似的化学性质。比较 LSD 和血清素时,观察到它们的最高占据分子轨道电子密度的相似性。氮原子由于其自由孤对电子而具有电子密度。

血清素

LSD

LSD 是通过使麦角酸与二乙酰胺反应制备的。LSD 的化学成分为 C20H25N3O。它有四种不同的异构体,但是只有当它为 R,R,立体化学时。只有一种立体异构体(d-)具有精神活性。因此,LSD 的外消旋混合物的效力只有右旋形式的一半。两种分子中吲哚环周围区域的电子密度最低。两种分子的偶极矩非常接近。血清素为 2.98 D,LSD 为 3.04 D;D 是电偶极矩的 SI 单位。偶极矩在两种分子中都指向胺基。两种分子几乎相同的偶极矩是 LSD 能够与血清素结合到相同受体的关键。
LSD 是一种相当稳定的有机分子,与具有相似结构的其他分子类似。主要需要注意的是水分,因为在水分存在的情况下会发生化学反应,还有氧气、光和温度。反应速率通常呈指数级地依赖于温度。
纯净的 LSD 是一种白色或透明、无味、水溶性的晶体,可以被碾碎成粉末并溶解。LSD 最常见的形式称为“吸墨纸酸”,即用 LSD 浸泡过的纸张。被称为“微点”的药片也很常见。LSD 通常以晶体形式存在,干燥在明胶纸上,或以胶囊或液体形式存在。制作 LSD 的方法有几种。一种常见的方法是从麦角酸开始。这将需要牵牛花种子。牵牛花及其种子含有麦角酸酰胺。它被认为是 LSD 的前体。不同种子的 LSA 含量各不相同。因此,用它制成的药物的质量也会不同。另一种合成路线是使用麦角菌。一旦获得麦角菌,就必须提取含有碱性氮原子的麦角碱。需要在暗室环境下工作,因为麦角菌在强光下会分解。LSD 本身在暴露于光线下时也会迅速分解。合成中使用的溶剂和试剂也有害。无水肼溶剂在加热时会爆炸。它是一种致癌物质。另一种经常用于合成的化学物质是氯仿。它也会致癌,同时也会对器官造成内部损伤。麦角碱通过加入试剂和加热合成成称为麦角酸异酰肼的化合物。麦角酸异酰肼被异构化以使其具有活性。冷却后,异构体与酸和碱混合,并蒸发。获得的产物是麦角酸二乙酰胺异构体,它再次被异构化以产生具有活性的 LSD。然后对 LSD 进行纯化和结晶,以获得更高浓度的形式。
没有关于 LSD 过量服用导致死亡的记录。它在生理上是耐受性良好的,并且没有证据表明对大脑或人体其他部位有持久性生理影响。LSD 可能会暂时损害做出合理判断和理解常见危险的能力,从而使使用者更容易发生事故和人身伤害。它可能导致短暂的混乱、抽象思维困难或记忆力和注意力集中力受损的迹象。
甲基苯丙胺是一种高度上瘾的药物和兴奋剂,可提高心率、血压和身体活动。它于 1919 年由日本发现,此前苯丙胺于 1887 年 1 月由罗马尼亚化学家拉扎尔·埃德莱安首次合成。1919 年甲基苯丙胺的首次使用是为了缓解疲劳和提高警觉性,随后被冠名为苯丙胺,一种吸入剂,在医学上用于治疗胸闷。甲基苯丙胺也称为冰毒、冰毒和冰,在二战期间被广泛用于保持清醒和提高警觉性。
化学 甲基苯丙胺的化学名称为 (S)-N,a-二甲基苯乙胺;d-N-甲基苯丙胺,化学式为 C10H15N。甲基苯丙胺是通过在苯丙胺的侧链上连接一个甲基来制成的,如图所示。甲基保护苯丙胺免受单胺氧化酶降解。由于单胺氧化酶无法降解甲基苯丙胺,因此它会持续存在于血液中,从而导致长期影响,如阿尔茨海默病、偏执狂和精神病行为。甲基苯丙胺具有两种异构体,因为甲基可以连接到苯丙胺的左侧或右侧,形成两个镜像。右旋甲基苯丙胺或右手甲基苯丙胺是两种异构体中活性更高、效力更强的一种。
{kind=link}
{kind=link}
{kind=link}
甲基苯丙胺的作用机制 甲基苯丙胺攻击神经递质,从而在体内产生依赖性。多巴胺、去甲肾上腺素和肾上腺素是甲基苯丙胺攻击的神经递质。多巴胺控制运动、愉悦、情绪和思维过程。当人类完成某件事时,大脑会释放多巴胺。多巴胺过多会让人产生欣快和幸福感。甲基苯丙胺的作用是刺激多巴胺释放,并阻断多巴胺的再摄取,从而在体内产生多巴胺浓度,如图所示。这种多巴胺浓度会让人产生兴奋和幸福感。当多巴胺的作用消失时,幸福感也会消失。这会导致上瘾和对欣快感的渴望,因此需要服用甲基苯丙胺。这种浓度会导致神经细胞死亡,从而产生长期影响,如精神分裂症、阿尔茨海默病和精神病行为。去甲肾上腺素负责控制警觉性、休息周期、注意力和记忆力。甲基苯丙胺也会阻断去甲肾上腺素的再摄取,从而让人更加警觉,不需要休息。肾上腺素的再摄取也会被甲基苯丙胺阻断,这也会产生浓度梯度,导致肾上腺素被吸收到大脑的不同部位,从而释放肾上腺素,并让人感到兴奋和激动。
{kind=link}
{kind=link}
影响 甲基苯丙胺的影响包括增加身体活动、减少食欲、警觉性、增加心率和血压、体温过高、偏执狂、意识混乱、焦虑、攻击性、失眠、震颤和易怒。
长期影响包括暴力行为、情绪障碍、妄想、精神病行为和幻觉、精神分裂症、中风和阿尔茨海默病。
概述
甲基苯丙胺通常简称为冰毒、冰、晶体或玻璃。它是一种具有高度上瘾性的精神活性药物。甲基苯丙胺可以吸烟、注射、鼻吸或口服。它曾被用作治疗多动症和肥胖症的低剂量药物。在高剂量下,甲基苯丙胺会导致欣快感和性欲亢进。长期使用甲基苯丙胺会导致脑损伤、心血管损伤和牙齿腐烂。
用途
甲基苯丙胺在医学上被用于治疗多动症和肥胖症。这是因为甲基苯丙胺的作用会导致能量和警觉性增强,以及食欲下降。该药物已获得 FDA 批准。另一方面,甲基苯丙胺在娱乐活动中被用于达到欣快感,因为它会导致大脑释放多巴胺。
效果
-立即影响:欣快感、性欲亢进、能量和意识增强、易怒、自信和暴力。常见的生理影响包括厌食、口干、头痛、恶心、腹泻、皮肤干燥、失眠和心律不规则。
-长期影响:对甲基苯丙胺的耐受性增加。牙齿腐烂和脱落。强烈的药物依赖性和渴望。药物性精神病。
-过量:可能导致心脏骤停或中风,并可能导致死亡。严重的幻觉和皮肤爬行的感觉也很常见。
-戒断:症状通常包括疲劳、抑郁、食欲增加和焦虑。影响通常持续数月。
化学概述
它的 IUPAC 名称为 N-甲基-1-苯丙烷-2-胺。它的化学组成是 C10H15N,分子量为 149.233 g/mol。甲基苯丙胺是苯乙胺家族的一员,该家族包括兴奋剂和致幻剂。甲基苯丙胺有两种对映异构体。S-异构体是正在描述的一种。它通常被制成粉末/晶体。甲基苯丙胺会导致多巴胺神经递质系统活性增加,从而产生这种欣快感。使用甲基苯丙胺后,多巴胺和血清素浓度会从使用前下降。
历史
甲基苯丙胺于 1893 年由化学家永井长吉首次发现和合成,并于 1919 年由绪方章首次结晶。
甲基苯丙胺在二战期间被广泛用于对抗盟军和轴心国军队士兵的疲劳和饥饿。它后来被 FDA 批准作为治疗嗜睡症、抑郁症、酒精中毒、肥胖症和多动症的药物。
[4] [5] [6] [7] 米诺环素 是一种四环类抗生素,可对抗体内的细菌。它用于治疗多种细菌感染,如泌尿道感染、呼吸道感染、皮肤感染、痤疮、淋病、衣原体感染等。

概述
[edit | edit source]米诺环素是一种口服抗生素,通常用于治疗寻常痤疮(俗称囊肿性痤疮)。米诺环素能够更有效地杀死痤疮细菌,并减少痤疮的红肿。它最常以米诺环素盐酸盐的形式分配,米诺环素盐酸盐是四环素的半合成衍生物。 [8] 该药物是作为以下抗生素销售的:米诺欣、丁那新、维克林、索洛丁和通用米诺环素。
它的 IUPAC 名称为 (4S,4aS,5aR,12aS)-4,7-双(二甲基氨基)-3,10,12,12a-四羟基-1,11-二氧代-1,4,4a,5,5a,6,11,12a-八氢四苯并 [a,c,g,i]环十二碳烯-2-酰胺 [9]
用法
[edit | edit source]米诺环素与其四环素类药物之一多西环素一起,是为痤疮患者开具的口服抗生素中最常见的形式。如果您对米诺环素或其他四环素类抗生素过敏,请勿服用。
有时它用于治疗莱姆病和其他皮肤感染,例如耐甲氧西林金黄色葡萄球菌 (MRSA),因为米诺环素的每天两次剂量比其他四环素类化合物所需的每天四次剂量更容易耐受。成人米诺环素的剂量为每天 100-200 毫克,但如果漏服剂量,可能会影响治疗过程。
如果漏服剂量,请不要加倍服用以弥补漏服的剂量。如果服用过量,请寻求紧急医疗救助或致电中毒控制中心,电话号码为 1-800-222-1222。
注意事项
[edit | edit source]如果怀孕,请不要使用米诺环素。它会导致胎儿受到伤害或永久性牙齿变色。米诺环素可能会降低避孕药的有效性。它还会通过母乳传递,影响哺乳婴儿的骨骼和牙齿发育。8 岁以下儿童不应服用米诺环素。服用米诺环素前不要服用维生素补充剂。这些补充剂会降低它的有效性。避免过度暴露在阳光下。服用米诺环素的人更容易晒伤。应按规定剂量服用米诺环素。任何少于规定的剂量都可能导致症状再次出现。如果超过有效期,请勿服用。过期的米诺环素可能导致肾脏损伤。
副作用
[edit | edit source]对阳光的敏感性(皮肤)(自动)
腹泻/恶心/胃部不适
头晕
嗜睡
头痛
呕吐
皮肤变色
自身免疫性疾病
胸痛/呼吸不规则
发烧(罕见)
皮肤/眼睛变色(罕见)
喉咙痛(罕见)
- ↑ http://publicsafety.utah.gov/investigations/methhistory.html
- ↑ http://methoide.fcm.arizona.edu/infocenter/index.cfm?stid=165
- ↑ http://www.montana.edu/wwwai/imsd/rezmeth/transmit.htm
- ↑ http://www.acnp.org/g4/GN401000166/Default.htm
- ↑ http://www.health.ny.gov/diseases/aids/harm_reduction/crystalmeth/docs/meth_literature_index.pdf
- ↑ http://chemistry.about.com/od/medicalhealth/a/crystalmeth.htm
- ↑ http://www.emcdda.europa.eu/publications/drug-profiles/methamphetamine
- ↑ http://www.rxlist.com/solodyn-drug.htm
- ↑ http://www.drugbank.ca/drugs/DB01017
- ↑ http://www.drugs.com/sfx/minocycline-side-effects.html
- ↑ http://en.wikipedia.org/wiki/Minocycline
5. "Drug Information Online". <http://www.drugs.com/minocycline.html>. 2012年10月28日
6. "Minocycline". <http://www.aocd.org/skin/dermatologic_diseases/minocycline.html>. 网页。 2012年10月28日。
7. "Minocycline for Acne." <http://minocyclineforacnereview.com/>. 网页。 2012年10月28日。 类固醇由许多类固醇环组成,这些环由“一个五碳环和三个六碳环”的组合组成(胆固醇是主要结构)(Kishner,第 1 页)。 类固醇包含从基本环结构获得的所有形式,其中之一是睾酮和合成代谢类固醇 (AASs)。

合成代谢类固醇是男性激素睾酮的合成版本,睾酮是男性特征的驱动力量,如肌肉生长、面部毛发、声音变低沉。 当身体吸收睾酮时,它会进入细胞的受体位点并激活细胞。 睾酮分子进入细胞后,由于 RNA 聚合酶的添加,蛋白质合成和磷酸盐合成显着增加。 磷酸盐合成允许生产额外的磷酸肌酸,这使得在没有氧气的情况下能够进行更多工作,而蛋白质合成增加则允许转录增加。 合成代谢类固醇还会导致体内氮保留增加。 身体需要正氮平衡来产生肌肉,因为当身体施加剧烈力量时会产生负氮平衡。 由于人体每天只产生大约 2-10 毫克的睾酮,而大多数 AAS 滥用者每周使用高达 1000 毫克的睾酮,因此它对肌肉生长的影响非常显着。

人体中主要的激素,它产生雄激素(一种由其他化合物组成的化合物),它管理不同男性特征的生长和滋养。 这是一个重要的激素,因为它有助于恢复组织的健康并提供肌肉质量(合成代谢:代表睾酮和二氢睾酮)。 因此,雄激素和合成代谢的双重作用使睾酮具有双重作用机制。
多年来,生物化学家一直在尝试转化和调整睾酮结构,以制造可以口服的药物,或者在溶解到体内后具有不同的分解方式,或者更好的是,一种可以同时做到两者的药物。 在经过多年的研究后,科学家得出结论,通过替代和改变睾酮分子中的某些部分,它可能会对效果产生很大影响; 但不幸的是,它尚未被发现。

睾酮和合成代谢类固醇已被用作治疗以下疾病的药物
- 多种类型的贫血
- HIV 消瘦综合征
- 骨质疏松症
- 严重烧伤
- 急性及慢性烧伤
- 营养不良、体重减轻
- 身材矮小
- 原发性或继发性性腺功能减退症
(斯蒂芬·基什纳,第 1 页)
虽然它被一些人正确使用,但许多其他人出于其他目的滥用该药物,例如为了增强他们正常的合成代谢和雄激素能力,以改善他们的身体形象和力量。


为了改进 AAS 的研究,生物化学家制定了一个目标,即改变分子,使其比睾酮“更合成代谢,更少雄激素”,这使得它可以口服,并减少对 HPG 轴的影响(斯蒂芬·基什纳,第 3 页)。 根据斯蒂芬·基什纳撰写的一篇名为“合成代谢类固醇的应用和滥用”的文章,“AASs 是从 3 种化合物中研发的:睾酮、二氢睾酮和 19-去甲睾酮”(Kishner,第 3 页)。(睾酮和 19-去甲睾酮是一种非常相似的化合物,非常相似,只是 19-去甲睾酮不包含第 19 个碳)。
对化合物进行的最早改变是在第 17 个碳上,通过添加甲基/乙基。 这种修饰扩展了药物的半衰期,因此允许药物保持活性并可以口服。

虽然这种修改在某些方面有所帮助,但它也产生了后果,因为这种新化合物与原化合物并不完全相同。 由于在化合物中添加了甲基/乙基,因此在转化过程中发现了肝脏染色。 此外,所有添加到碳-17 的化合物都受到影响,并对肝脏造成化学损伤。(需要来源)

- 中枢神经系统:增加性欲、幸福感、攻击性、空间认知
- 下丘脑/垂体:降低 GnRH、LH、FSH,增加 GH
- 肝脏:降低 SHBG、HDL
- 喉:降低声音
- 乳房发育
- 肾脏:增加红细胞生成素
生殖器:增加发育、精子生成、勃起
- 前列腺:增加大小、分泌
- 皮肤:增加面部/体毛、皮脂分泌
- 骨骼:增加 BMD
- 肌肉:增加瘦肉质量、力量
- 脂肪组织:增加脂肪分解,减少腹部脂肪
- 血液:增加红细胞压积
- 免疫系统:增加自身抗体产生
Kishner, Stephen. "Anabolic Steroid Use and Abuse." Medscape. Emedicine, 2011 年 7 月 6 日。网页。 2011 年 12 月 13 日。
Auchus, Richard J. MD, PhD. "The Science Of Anabolic Steroid Abuse." Utsoutwestern.edu. 网页。 2012 年 10 月 20 日
http://www.vanderbilt.edu/AnS/psychology/health_psychology/anabolic_steroids.html
图片:维基媒体公用 羟考酮是一种止痛药,口服药物,用于缓解中度至重度疼痛。 它于 1916 年在德国被开发出来,旨在改进现有的阿片类药物,阿片类药物是从鸦片罂粟中提取并用于治疗目的的药物。 常见的复方药物包括布洛芬等非处方止痛药和羟考酮与对乙酰氨基酚的复方。 许多品牌名称包括 OxyContin,这是辉瑞制药公司生产的缓释口服羟考酮变种品牌,Roxycodone 和 Xanodyne。

羟考酮主要用于缓解中度至重度疼痛,以及管理急性慢性疼痛。 它还可以用作严重腹泻和肠易激综合征的替代药物,当常用的药物效果不佳时。 羟考酮比吗啡引起更少的镇静、呼吸窘迫、瘙痒和恶心,因此身体更容易耐受羟考酮而不是吗啡。 如果羟考酮作为复方制剂服用,请务必阅读有关成分的所有信息,并咨询您的医生和药剂师以获取更多有关安全用药的信息。
最常报告的负面副作用包括令人不安的噩梦、记忆力减退、疲劳、头晕、便秘、情绪变化、潮红、头昏眼花、头痛、口干、瘙痒、出汗过多、焦虑和视力下降。少数患者出现腹痛、腹泻、食欲不振、尿潴留、呼吸困难和阳痿。过量服用会导致呼吸浅表、皮肤湿冷、低血压、瞳孔缩小、呼吸停止和死亡。

剂量和给药
[edit | edit source]羟考酮有多种形式:溶液(包括浓缩液)、片剂、胶囊和缓释/长效。前面的羟考酮形式列表通常在饭前或饭后每 4 到 6 小时服用一次,具体取决于药物是用于缓解暂时性疼痛还是作为定期服用的药物。请务必遵循处方标签上的说明,并向医生或药剂师咨询您不完全理解的任何信息。
如前所述,它可以通过口服或鼻腔,使用静脉注射或直肠给药。所有羟考酮使用量中,60-87% 是通过口服给药,其余部分则为口服给药。与吗啡相比,口服给药的效力高出 1.5 到 2 倍。由于其效力,医生可能会让患者从低剂量开始,并在疼痛无法控制的情况下随着时间的推移逐渐增加剂量。身体可能会对药物产生耐受性,在这种情况下,医生可能需要增加剂量。
这种药物有成瘾性,因此患者不得服用过量。患者也不得比医生规定的更频繁地服用羟考酮,也不得比医生推荐的时间更长时间地服用羟考酮。不要突然停止服用药物;你可能会出现戒断症状。
戒断
[edit | edit source]当患者突然停止服用药物时,他可能会出现焦虑、惊恐发作、失眠、肌肉疼痛、恶心、发烧、流感样症状(流鼻涕、打喷嚏、发冷、流泪等)和肌肉无力。当出现戒断症状时,医生通常会逐渐减少剂量。大多数严重戒断病例与那些上瘾并非法使用羟考酮的人有关。由于代谢等方面的差异,每个人从羟考酮中戒断的经历都是独一无二的。因此,即使是相同剂量的药物,一个人可能会比另一个人经历更严重的戒断症状。通过适当的排毒程序可以减轻戒断症状。[2]
药物滥用
[edit | edit source]羟考酮是一种强效药物,青少年和成年人都会使用。这种药物让使用者处于兴奋状态。由于它是一种止痛药,它在进入血液后会让使用者感到平静放松几个小时。随着使用者越来越频繁地服用这种药物,对其的耐受性会越来越高。当上瘾变得迫在眉睫时,某些副作用可能会开始出现,并毁掉药物滥用者的社交生活。上瘾带来的某些影响包括:不断地想着羟考酮,获取大量这种药物,偷偷地服用这种药物,不让别人知道,对亲人撒谎,以及在夜间睡眠时感到疼痛和焦虑。由于羟考酮含有对乙酰氨基酚,服用过量的药物会导致以后出现肝脏问题,有时甚至会致命。
特殊注意事项
[edit | edit source]羟考酮是一种敏感且有效的药物,因此在服用之前需要注意一些事项。请告知您的医生和药剂师
- 如果您对某些药物过敏,例如可待因、希考丹和其他任何药物
- 如果您正在服用其他药物(包括维生素或营养补充剂),或者您正在服用或计划服用其他药物。
- 如果您现在或曾经患有哮喘、肺病或麻痹性肠梗阻
- 如果您一直在喝酒或曾经大量喝酒,以及如果您使用过任何街头毒品(包括药物过量)
- 如果您怀孕或计划怀孕,或者如果您正在哺乳
- 如果您即将进行手术(包括牙科手术)
品牌名称
[edit | edit source]羟考酮也称为
- Dazidox
- Endocodone
- Oxy IR
- ETH-Oxydose
- Oxyfast
- Roxicodone
- Oxycontin
- Oxexta
参考文献
[edit | edit source]- "Oxycodone." PubMed Health. October 15, 2011. AHFS Consumer Medication Information. October 26, 2012 <http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000589/>
- "Oxycodone." Wikipedia. October 24, 2012. October 26, 2012 <http://en.wikipedia.org/wiki/Oxycotton#Medical_uses>
"Oxycodone" WEBMD. November 20, 2012 http://www.webmd.com/drugs/mono-5278-OXYCODONE+-+ORAL.aspx?drugid=1025&drugname=oxycodone+Oral&source=1
简介
[edit | edit source]避孕,也称为节育,是一种家庭计划的形式。这种方法可以预防意外怀孕,有时还可以预防性传播疾病。某些形式的避孕方法仅供男性使用,而其他形式的避孕方法仅供女性使用。
其他家庭计划技术包括节欲、手术绝育、自然家族计划和体外射精。
屏障方法
[edit | edit source]避孕屏障方法是指使用物理或化学形式来阻止精子进入子宫,从而防止卵子受精,进而避免怀孕。除了对装置内材料的过敏反应外,它们没有明显的副作用,并且可以在性交前使用。
男性避孕套:这是现代世界上最常见的避孕形式。男性避孕套由男性在任何类型的性交前将薄管卷到勃起的阴茎上使用。它在 20 世纪成为一种非常流行的避孕方法,并且是防止大多数性传播疾病的最有效避孕形式,前提是只使用一次。最早的男性避孕套是用动物产品制成的,如今在美国市场上被认为是高端产品。如今最常见的一种避孕套是乳胶橡胶避孕套,有润滑和不润滑两种类型。
女性避孕套:这种避孕套看起来与男性避孕套非常相似,但实际上是插入女性体内,而不是男性。它是一种一次性使用、预先润滑的聚氨酯套,也用于性交前。它有两个环——一个插入阴道上端,另一个留在阴道外,覆盖阴唇和阴道周围区域。由于材料成本高昂,它并没有得到广泛使用。
杀精剂:杀精剂是用于杀死精子的化学物质,因此,它们不允许精子进入阴道并导致怀孕。在某些情况下,它们甚至可以杀死某些性传播疾病。它们以凝胶、凝胶剂、泡沫、栓剂和水溶性薄膜的形式出售。杀精剂通常与其他类型的屏障方法一起使用,少量使用是安全的,因为高剂量会导致阴道内膜损伤,阴茎敏感,甚至增加感染艾滋病毒的可能性。
海绵:海绵有不同类型和尺寸。它通常由一次性使用和柔软的聚氨酯制成,并配有杀精剂。这是女性的一种避孕选择,因为它非常方便——女性只需用水浸湿海绵,将其插入阴道,它就可以在接下来的 24 小时内有效,无论性交次数多少。尽管很方便,但它仍然不如其他避孕方法有效。
子宫帽:橡胶、金属或乳胶子宫帽是一种小型、杯状的工具,放置在阴道内的子宫颈上。可以在圆顶内放置杀精剂,以确保精子在物理和化学上都被阻止。某些类型的子宫帽必须与阴道壁产生吸力,才能在性交过程中保持在适当位置。由于每个子宫颈都不一样,每个女性都必须由医疗专业人员正确地进行测量。很少有女性实际使用这种屏障避孕方法,因为没有多少专业人员接受过子宫帽测量的培训。在美国,食品药品监督管理局 (FDA) 批准了三种类型的子宫帽——小型、中型和大型。小型子宫帽专为从未怀孕的女性设计,中型子宫帽专为流产、堕胎或剖腹产的女性设计,大型子宫帽专为顺产的女性设计。
隔膜:与子宫帽一样,隔膜也是一种用于子宫颈的避孕屏障。这种圆顶形装置可以用乳胶或硅胶制成,并在一个环上扩展。环的边缘有一个弹簧,它也会在阴道上产生吸力。就像子宫帽一样,隔膜也需要根据子宫颈的大小进行测量。这种方法的缺点是,隔膜必须在性交前 3 小时放置。
避孕的激素方法通过操纵卵巢激素(如孕激素和雌激素)来阻止卵子的受精和排卵。这些方法包括口服避孕药、宫内节育器、注射剂、植入剂、避孕贴和阴道避孕环。虽然这些避孕方式确实有助于防止意外怀孕,但它们不能预防性传播疾病。
联合口服避孕药:这些避孕药以药丸形式提供,必须每天服用一次。药丸会阻碍垂体中的促卵泡激素 (FSH),从而阻止卵巢中卵泡的成熟。通过人体的反馈回路,黄体生成激素 (LH) 会增加,这会阻止卵巢内的卵子排卵。除了减少排卵外,药丸中孕激素的增加还会增加宫颈粘液,从而为精子创造一个恶劣的环境。两种类型的避孕药包括联合孕激素和雌激素药丸以及只含孕激素的药丸。通常,女性需要口服 20、21 或 22 片活性药丸,其中包含一定量的联合孕激素和雌激素,然后服用非活性糖丸。在服用非活性药丸期间,女性的月经量会比没有服用药丸时减少。这些药丸模拟了正常但有规律的月经系统。联合口服避孕药的例子包括 Yaz、Yazmin、Ortho Tri-Cyclen、Desogen。
连续使用口服避孕药:这些药丸也必须每天口服,并包含孕激素和雌激素的组合。这些药丸的目的是减少一年中月经周期的次数和持续时间。使用连续使用口服避孕药,女性需要连续服用 12 周的活性药丸,然后服用 7 片非活性药丸。这种方法的缺点是女性会出现突破性出血和斑点。这种类型的避孕药的一个例子包括 Seasonique。
只含孕激素的药丸:这些药丸只包含孕激素,以增加宫颈粘液的厚度并减少精子的渗透。这种方法也使用每天一次的药丸,就像联合口服避孕药和连续使用口服避孕药一样。许多提供者选择为那些对联合药丸中的雌激素出现不良副作用的女性开这种避孕药。只含孕激素的药丸不如联合药丸有效。
紧急(性交后)避孕:这种避孕方法是口服的,并且只在女性发生可能导致意外怀孕的无保护性交后立即(大约 24-36 小时)服用一次。然而,它在无保护性交后五天内可能仍然有效。这种“事后避孕药”每片只含有 750 毫克的左炔诺孕酮。它只是一种一次性使用,而不是持续性使用。
宫内节育器(IUD)是放置在子宫颈内的塑料装置,可能含有铜和/或释放左炔诺孕酮,用于阻止精子到达已植入的卵子。早期的 IUD 版本有一个铜质分支,用于将其固定在阴道内。随着更现代的塑料变体的出现,人们观察到,随着装置尺寸的增加,月经量会增加,如果女性与患有性病的人发生性接触,她们更容易患上阴道感染。对于这种类型的避孕方法,许多风险似乎超过了优势。
注射剂是口服避孕药的持久且更有效的版本。注射剂以每月一次的肌肉注射形式提供给女性,使用高剂量的醋酸甲羟孕酮 (DMPA),也称为 Depo-Provera,以及以每两个月一次的肌肉注射形式提供,使用炔诺酮烯安酯 (NET-OEN)。使用注射剂作为避孕方式的唯一负面方面是如何在很长一段时间内保持规律的剂量。
预计未来注射剂的使用将比其他避孕方法更普遍。只含孕激素的注射剂很可能会成为最常用的和提供的避孕方法,例如每月一次的 DMPA 和每两个月一次的 NET-EN。就联合注射剂而言,可以提供每月一次的剂量。正在改进一种由炔诺孕酮丁酸酯制成的每三个月一次的注射剂,其作用机制类似于 DMPA。对于每三个月一次的剂量,例如 DMPA,最大的优势在于体内循环的合成化学物质含量低,女性更容易更快地停止服用避孕药以再次生育。此外,较少的激素化学物质会导致卵巢限制减少,并且出现闭经(一个或多个月经周期的缺失或错过)的风险降低。
Depo-Provera 是一种长效激素注射避孕方法,在避孕目的方面非常成功,但很难从体内清除,并且需要更长的时间才能再次怀孕。醋酸炔诺酮烯安酯是 Depo-Provera 的一种减少孕激素的版本,它可以稍微缓解 Depo-Provera 的不良副作用,但女性需要每两个月注射一次。
注射剂作为长效避孕方式的副作用包括子宫肌瘤增大、闭经、高血压、糖尿病、排卵延迟恢复等。在某些情况下,持续使用注射剂与骨质疏松症有关。这些可注射制剂持续时间很长,如果患者想停止这种避孕方式,可能需要数周甚至数月才能恢复生育能力。Depo-Provera 的主要副作用是随着时间的推移骨密度下降。建议患者摄入钙补充剂以抵消这种副作用。Depo-Provera 不建议使用超过两年。
这些植入剂提供了一种持久且持续的避孕方法。植入剂必须通过手术植入皮下,也必须从同一区域取出。就其结构而言,植入剂通常在胶囊或棒状物中放入类固醇。由于这种形式提供了稳定的孕激素剂量,植入剂通常包含比注射剂和口服避孕药更少的激素。手术植入和取出通常需要专业人士,取出时间比植入时间更长。
http://www.americanpregnancy.org/preventingpregnancy/overviewtypesbirthcontrol.html
Senanayake, P. (2008). Atlas of Contraception (第 2 版). 英国:Informa UK Ltd。
吗啡源自鸦片罂粟 Papaver Somniferum 的汁液,约占植物汁液的 10%,已被用于缓解各种类型和严重程度的疼痛数千年。在 1806 年被分离出来之前,它被作为称为鸦片的干汁液摄取。无论是鸦片还是单独使用,注射、吸烟或吞服,吗啡都会与大脑中的受体结合,产生欣快感,使其在娱乐用途上很受欢迎。在肠道中,它与相同的受体结合,并能很好地减缓蠕动运动,使其成为治疗腹泻的无与伦比的药物。与任何阿片类药物一样,长期使用者最终会成瘾,并且耐受性——需要更高的剂量才能达到相同的效果——可以预期会出现在镇痛(止痛)和吗啡产生的欣快感(高潮)中。
虽然作为对抗疼痛的武器必不可少,但缓解会伴随一些可能并不受欢迎的“副作用”,特别是嗜睡、视力模糊、便秘和食欲下降。当然,吗啡引起身体依赖性(“成瘾”)的可能性是其最糟糕的不良影响,因为如果这个人突然停止或大幅减少吗啡的剂量,戒断症状会非常令人不快。


吗啡可以以多种方式给药:静脉滴注、口服药丸、注射、鼻腔(吸入)或直肠;每种方法的有效程度各不相同,口服药丸的有效程度最低(10-30%),静脉滴注的有效程度最高(接近 100%)。它的作用机制是与大脑和脊髓(主要位于感觉疼痛的第一个处理部位——胶质层)中的 μ-阿片受体、κ-阿片受体和伤害感受素受体以及大脑中的 δ-受体结合。它抑制了来自周围神经系统中疼痛神经元的信号的传递,并且还会在背角产生影响以兴奋神经元和其他通路。结果是吗啡阻止了来自中枢神经系统和周围神经系统的疼痛信号。具有讽刺意味的是,这种药物并没有阻止疼痛的传递,而是改变了使用者对疼痛的感知;他们意识到了疼痛,但发现它是可以控制的,或者可以忽略。
只需 5 毫克吗啡即可产生明显的影响,因为该药物与人体自然产生的内啡肽、脑啡肽和强啡肽结合到相同的受体上,这些物质都是类似阿片的物质。
吗啡产生的欣快感是另一种机制的一部分,这种机制涉及 γ-氨基丁酸 (GABA) 抑制剂及其各自的神经元。在细胞条件下,GABA 会减少大脑中多巴胺的释放量,多巴胺是一种与快乐相关的脑神经递质。吗啡会抑制大脑中 GABA 的释放量。随着时间的推移,它会逐渐增加大脑中的多巴胺水平,从而导致“兴奋”或欣快感。
此外,长期使用吗啡会抑制环磷酸腺苷 (cAMP) 的产生。当吗啡突然变得不可用时,人体会产生更多的 cAMP,从而导致过度活跃和药物渴望。
不良反应
[edit | edit source]上瘾
[edit | edit source]吗啡的使用以引起“上瘾”——身体依赖而闻名。心理上瘾也是可能的,但这是一个不同的问题,其原因不如身体依赖(尤其是对吗啡的依赖)来得那么具体,身体依赖是一种更容易理解的现象。
身体依赖指的是,当一定剂量成为每天必须服用才能避免不愉快的“戒断综合征”症状的必需品时,这是一种使用吗啡后几乎所有服用过它的人都会出现的情况。这需要多长时间在相当窄的范围内有所不同,但在大多数人通常服用一周或十天内不太可能出现。在此之后,应与医生讨论,以防止因未经治疗的戒断或因害怕“上瘾”而无故停止止痛治疗而导致不必要的痛苦。
其他阿片类药物也会导致身体依赖,尽管在戒断的严重程度和对药物耐受性的发展方面有所不同。
便秘
[edit | edit source]作为一种阿片类药物,吗啡有作用于肌间神经丛的趋势,肌间神经丛位于肠道中。这导致肠道动力下降,从而导致整体便秘效果。吗啡通过抑制肠道蠕动过程来降低肠道转运速度。
耐受性
[edit | edit source]对吗啡的耐受性很容易产生,这意味着需要更高浓度的吗啡才能获得相同的感觉或效果。关于这种耐受性如何产生的假设包括受体构象磷酸化的改变、受体与 G 蛋白解偶联、cAMP 通路的向上调节以及 μ-阿片受体内化。
戒断
[edit | edit source]吗啡戒断按以下方式进行,大致按照症状、发病时间和持续时间进行分类
- 第一阶段:强烈渴望缺失的药物(“渴望”)和焦虑;在最后一次服药后 6 到 12 小时开始。
- 第二阶段:除了渴望和焦虑(可能会变得更强)外,典型的症状包括出汗、流鼻涕、打哈欠和流泪(“流泪”)。通常在最后一次服药后约 8 到 12 小时内开始。
- 第三阶段:全身感到不适、全身酸痛和食欲不振是典型的症状发展。此时,肌肉痉挛(尤其是腿部痉挛)是“踢”一词的来源(最初用来描述鸦片戒断,后来用来描述放弃任何其他习惯),以及腹部痉挛是接下来出现的症状,大约在最后一次服药后 24 小时出现。
- 第四阶段:这是急性阶段,此时症状处于最严重状态,可能持续两到三天,然后消退。疼痛持续存在,尤其是在关节处。失眠、恶心、呕吐、腹泻是典型的症状,尽管它们差异很大——个人之间以及“戒断”尝试之间差异很大。男性有时会无意识地射精。严重的抑郁也是一种非常常见的症状。
致命组合/预防措施
[edit | edit source]吗啡是一种潜在的危险药物,因为它除了止痛(镇痛)外还会抑制呼吸。这种影响随着剂量的增加而增加,但也会受到耐受性的影响,因此只有在过量服用或与某些抑制剂(如酒精)一起服用时才会成为危险。
参考文献
[edit | edit source]"THE BRAIN FROM TOP TO BOTTOM." THE BRAIN FROM TOP TO BOTTOM. 道格拉斯医院研究中心,n.d. 网页。2012 年 10 月 28 日。<http://thebrain.mcgill.ca/flash/i/i_03/i_03_m/i_03_m_par/i_03_m_par_heroine.html>.
"吗啡." 药物论坛。网页。2012 年 10 月 28 日。<http://www.drugs-forum.com/forum/showwiki.php?title=Morphine>.
"吗啡." 来自 Drugs.com 的信息。N.p.,n.d. 网页。2012 年 10 月 28 日。<http://www.drugs.com/morphine.html>.
"吗啡." 吗啡。N.p.,n.d. 网页。2012 年 10 月 28 日。<http://www.emsb.qc.ca/laurenhill/science/morphine.html>.
# "Morphine Addiction Withdrawal Symptoms and Treatment" <http://www.rehabinfo.net/morphine-addiction/#stages>
简史
[edit | edit source]许多人听说过乳镁片,它是一种乳白色的产品,用作抗酸剂,以抵消胃酸过多引起的不适。但有多少人听说过乳忘片?这是有些人用来描述称为丙泊酚或地西泮的静脉麻醉剂的术语。
生产该药物的公司
[edit | edit source]该药物的生产商是阿斯利康。这家公司是瑞典阿斯利康 AB 与英国善宁集团 PLC 合并的结果。新合并后的公司于 1999 年 4 月 6 日开始运营。这两家公司希望将他们的努力合并成一家公司,以便他们能够作为一家生物制药公司在研发方面取得更大的进步。
关于该药物
[edit | edit source]丙泊酚是一种液体药物,通过静脉注射给药。它是一种全身麻醉剂,通过减缓大脑和神经系统的活动来启动和维持麻醉。它通常用于镇静接受过人工呼吸的患者或即将进行手术的患者。剂量可能因所需效果和患者年龄而异。55 岁以上的老年患者最初接受 20 毫克的剂量,然后维持在 0.05-0.1 毫克/公斤/分钟静脉注射。相比之下,55 岁以下的年轻患者最初接受 40 毫克的剂量,然后维持在 0.1-0.2 毫克/公斤/分钟静脉注射。丙泊酚也用于儿科和重症监护病房的患者,剂量会相应调整。
医疗用途
[edit | edit source]丙泊酚用于诱导和维持麻醉,在很大程度上取代了硫喷妥钠用于此适应症。丙泊酚也用于镇静接受机械通气的患者。在危重患者中,丙泊酚已被发现优于劳拉西泮,无论是在有效性方面还是在总体成本方面。丙泊酚也用于镇静,例如在内窥镜检查期间。与咪达唑仑相比,这种药物在这些环境中的使用会导致更快的恢复。
药物用途
[edit | edit source]任何对该药物中任何成分过敏或对鸡蛋、蛋制品、大豆或大豆制品过敏的个人应避免使用该药物。
某些医疗状况可能会与该药物相互作用。如果任何医疗状况适用于他们,个人应告诉他们的医生
- 服用任何处方药或非处方药、草药制剂或膳食补充剂
- 怀孕、计划怀孕或正在母乳喂养
- 患有胰腺炎、高血脂或癫痫
注意:苯二氮卓类药物、麻醉止痛药或其他镇静剂等药物会与这种药物相互作用,并可能增加这种药物副作用的风险。
优点
[edit | edit source]丙泊酚起效快,持续时间短,这意味着患者可以更快地恢复日常活动和饮食。其他药物会迫使患者花更多时间完全康复,才能恢复到基线活动。丙泊酚的另一个优势是可以替代阿片类药物。阿片类药物通过与人体中枢和周围神经系统中三个受体位点之一结合发挥作用。阿片类药物都有镇静作用,但通常伴随着恶心和呕吐等副作用。通过用丙泊酚替代阿片类药物,医生可以减少患者出现的这些副作用。
尽管丙泊酚有很多优点,但也要注意该药物的缺点。与吗啡或咪达唑仑等药物不同,Diprivan 没有可以逆转其作用的药物。任何药物的作用都必须持续到药物完全代谢并通过尿液从体内排出为止。与许多药物一样,患者对不同药物的反应也不同。不到一半服用药物的人对药物的反应如预期,因为基因的微小差异会改变他们身体对药物的反应方式。更具体地说,制造细胞色素 P450 蛋白的基因的微小差异是罪魁祸首,因为这些蛋白质是处理许多药物的蛋白质。这激发了人们对创建针对特定个体的“个性化药物”的兴趣。在此之前,丙泊酚仍然存在风险,例如会导致呼吸正常的患者毫无征兆地进入完全呼吸停止状态。
保持注射部位清洁至关重要,以防止细菌感染的风险。根据 RxList 的说法,Diprivan 中含有少量的依地酸二钠,以帮助防止细菌等微生物的生长。这也是 Diprivan 以单次使用包装出售的原因,以防止打开的药瓶中滋生这些微生物。科学研究表明,药物污染会导致发烧、感染甚至死亡。Diprivan 通常通过以下三种方式之一给药:注射器、输液和容积泵。
根据词典定义,注射器是“一种带有空心针头的管子,用于注射或抽取液体”。这种给药方法不言而喻。
输液泵将 Diprivan 等药物静脉注射到患者体内。这些通常在医院的病房里看到,在那里有一个液体袋挂在杆子上,以医生确定的速度将静脉输液滴入患者体内,以确保剂量和效果准确。
容积泵是一种类似于输液的给药方法,只是它们通常用于需要将药物或液体长期输注到系统中的长期护理患者。
"Diprivan." RxList. N.p., 25 Oct. 2010. Web. 20 Nov. 2012. <http://www.rxlist.com/diprivan-drug.htm>.
"Diprivan (Propofol) ." AstraZeneca. N.p., n.d. Web. 20 Nov. 2012. <http://www.astrazeneca.com/Medicines/Neuroscience/Product/Diprivan>.
Euliano, T. Y., and J. S. Gravenstein. "A brief pharmacology related to anesthesia." Essential Anesthesia: From Science to Practice. Cambridge, UK: Cambridge University Press, 2004. 173. Print.
"History." AstraZeneca. N.p., n.d. Web. 20 Nov. 2012. <http://www.astrazeneca.com/About-Us/History>.
"Propofol Sedation: Who Should Administer?." Institute for Safe Medication Practices. N.p., 3 Nov. 2005. Web. 20 Nov. 2012. <http://www.ismp.org/newsletters/acutecare/articles/20051103.asp>.
"The New Genetics." National Institute of General Medical Sciences 7.662 (2006): 64. National Institutes of Health. Web. 20 Nov. 2012.
寻找癌症的治愈方法一直是许多医护人员的目标。许多人尝试过,但很少有人取得像安东尼奥·格里洛-洛佩斯博士那样大的进步。他与几位同事一起开创了一种名为利妥昔单抗的新药,该药是第一个获得 FDA 批准用于治疗癌症的抗体。根据癌症的严重程度,这种药物可以完全治疗或延长患者的寿命。这一非凡的壮举有着非常简陋的起源。
格里洛-洛佩斯博士的根源在波多黎各。在成长过程中,他在波多黎各大学圣胡安分校获得了理学学士学位和医学博士学位。1980 年至 1990 年,他曾在密歇根大学担任副教授。他也是杜邦-默克制药公司帕克戴维斯公司的成员。后来,他被一家当时名为 IDEC 的初创公司提供了一个职位。格里洛-洛佩斯博士于 1992 年 11 月至 2001 年 1 月在该公司担任首席医疗官。后来,他于 2001 年 1 月至 2003 年 11 月担任首席医疗官名誉职位。正是在这家公司里,利妥昔单抗诞生了。
利妥昔单抗是一种液体药物,通过静脉注射到患者体内。它用于治疗和护理非霍奇金淋巴瘤、慢性淋巴细胞白血病、类风湿性关节炎、肉芽肿性血管炎和显微镜下多血管炎患者。剂量可能因所需治疗而异。例如,在类风湿性关节炎患者中,它以两周间隔两次剂量给药。但在非霍奇金淋巴瘤中,它每周一次,持续四到八周。在美国,它的商品名为利妥昔。在欧洲,它被称为 MabThera。主要关注的是非霍奇金淋巴瘤,即 NHL。
NHL 是一个包含除霍奇金淋巴瘤以外所有淋巴瘤的术语。霍奇金淋巴瘤的特点是存在称为瑞氏-斯腾伯格细胞的双核巨细胞。淋巴瘤是血液中淋巴细胞的癌症。淋巴细胞是一种白细胞,在人体免疫系统中起着不可或缺的作用。它们分为 B 细胞、T 细胞和自然杀伤细胞。在 NHL 患者中,B 细胞上发现了一种称为 CD20 的抗原。利妥昔单抗抗体与 CD20 抗原结合,从而破坏由于疾病导致的过量、过度活跃或功能失调的 B 细胞。
利妥昔单抗通过三种主要机制杀死癌性B细胞:补体依赖性细胞毒性 (CDC)、抗体依赖性细胞介导的细胞毒性 (ADCC) 和凋亡。CDC发生在大量血浆蛋白 (补体) 协同作用破坏入侵病原体和恶性细胞时。抗体-抗原复合物,例如利妥昔单抗和CD20之间的复合物,会激活补体。补体蛋白C1以“锁和钥匙”的方式与利妥昔单抗的尾部结合,并启动一系列反应,在B细胞膜上形成膜攻击复合物,进而形成孔隙,使细胞内容物流出并最终死亡。ADCC是一个过程,其中抗体-抗原复合物形成并吸引免疫系统的其他成分,包括自然杀伤细胞。这些细胞上的受体识别并与利妥昔单抗的尾部结合。自然杀伤细胞还携带充满细胞毒性分子的颗粒。当自然杀伤细胞与利妥昔单抗结合后释放颗粒时,颗粒会穿透B细胞的细胞膜,造成孔隙,促进细胞内容物释放,导致细胞死亡。颗粒还可以通过攻击细胞核来破坏细胞。最后一种机制被称为凋亡或程序性细胞死亡。凋亡被定义为细胞死亡,发生在生物体生长或发育的正常和受控的一部分。当利妥昔单抗与CD20结合并形成抗体受体复合物时,它会向细胞发出启动该过程的信号。细胞骨架向内坍塌,细胞核浓缩,DNA通过酶片段化为小片段。膜结合的囊泡也被破坏。在凋亡结束时,细胞已有效地自我毁灭。然而,目前尚不清楚这些机制是独立起作用还是协同作用。尽管如此,利妥昔单抗仍然被证明是一种有效的治疗方法。
相关实验
[edit | edit source]利妥昔单抗是在多个涉及小鼠的实验后产生的。三只小鼠被注射了来自淋巴瘤肿瘤的细胞。这些恶性细胞携带仅存在于B细胞上的CD20抗原。观察表明,小鼠的免疫系统产生了抗体来对抗这些外来物质。抗体的尾部是人源化的。将小鼠和人类基因序列结合使用,创造了嵌合抗体,即利妥昔单抗。
通往成功的道路
[edit | edit source]格里洛-洛佩斯博士将他的大部分成功归功于1908年诺贝尔奖得主保罗·埃尔利希。已故的保罗·埃尔利希是一位德国科学家,在血液学(血液生理学研究)和免疫学领域都有经验。他在后一个领域的成就使他于1908年与伊利亚·伊里奇·梅奇尼科夫共同获得诺贝尔生理学或医学奖。埃尔利希普及了“魔弹”药物的概念。这意味着药物能够选择性地靶向致病微生物。这样,就可以将毒素专门传递到该微生物。这是格里洛-洛佩斯博士工作的基础。利妥昔单抗通过选择性地与CD20抗原结合,形成抗体受体复合物,这是破坏受感染B细胞所用机制的关键部分。埃尔利希说过,研究的成功只有在几个大“G”的帮助下才能实现。在德语中,这些分别是Geduld、Geschicklichkeit、Glück、Geld和Geräte。它们分别翻译为坚持和决心、技能、运气、金钱和工具。这种疗法批准过程背后的故事表明了埃尔利希对格里洛-洛佩斯博士的影响。
道路上的障碍
[edit | edit source]美国食品药品监督管理局 (FDA) 负责通过颁布和执行有关食品和药品等产品的法律法规来保护公众健康。所有药物都必须通过他们的批准程序。安东尼奥·格里洛-洛佩斯博士将其比作蜗牛的速度,并认为它们是“及时开发和批准抗癌药物的最大障碍”。他举了几个例子,比如仅仅为了批准利妥昔单抗的拟议名称而进行了为期五个月的审查。PanThera没有获得批准,不得不修改为美国市场上的Rituxan和欧洲市场上的MabThera。在数字时代之前,电子书和社交网站不可用,一切都必须手工完成。格里洛-洛佩斯博士和他的IDEC同事切特·瓦恩斯、艾丽丝·魏和约翰·莱昂纳德不得不将一箱又一箱的材料送到FDA,以供他们审查并批准利妥昔单抗的使用。在数据集中,有一个患者IDEC小组认为由于一些小错误而无法评估。为了阻碍审批过程,FDA的一位医学审查员要求他们考虑将该患者作为可评估患者。这有利于药物制造者,因为这位特定患者对药物产生了完全的反应。这提高了总反应率 (ORR) 和完全反应率 (CR)。该团队提交了他们的生物制剂许可申请,并在缓慢的九个月后,于1997年11月26日最终获得批准。此后,格里洛-洛佩斯博士及其同事仍然面临一些障碍,例如人们怀疑使用利妥昔单抗作为R+CHOP联合方案的一部分,而不是传统的方法(在化疗后使用抗体,而不是联合使用)是否有效。尽管存在这些疑虑,但联合方案被证明是非霍奇金淋巴瘤的治疗方法。格里洛-洛佩斯博士按照保罗·埃尔利希的理念,坚持不懈,最终成功找到了治疗癌症的有效方法。
最终结果
[edit | edit source]在开发过程中,取得了一些里程碑。临床试验由自愿参与的患者以创纪录的速度完成,他们甚至会打扮成老鼠,回想起这种抗体的嵌合性质。利妥昔单抗成为第一个获得FDA批准用于治疗癌症的抗体。格里洛-洛佩斯博士甚至为更快的FDA审批铺平了道路。以前,FDA平均需要七年才能批准一种许多患者迫切需要的药物。他能够将审批时间缩短了一半。2004年Discovery Health Channel的医学荣誉表彰了一些成就。利妥昔单抗的开发不依赖于政府支持。投资者和合作伙伴为整个项目提供资金。没有联邦或州拨款、激励措施或研究资金。
影响
[edit | edit source]利妥昔单抗的影响是前所未有的。它为可治愈的淋巴瘤(如弥漫性大B细胞淋巴瘤)提供了治疗方法。不可治愈淋巴瘤患者的寿命得到了延长。最近的数据进一步证明了利妥昔单抗的成功。自2001年以来,它一直被认为是世界上最有效的抗癌药物。仅2010年的销量就达到67亿美元。每年约有50,000名淋巴瘤患者被治愈。自1997年上市至2010年,已有超过200万患者接受了治疗。在此发现之前,在寻找治疗方法或延长寿命方面一直处于停滞状态。希望安东尼奥·格里洛-洛佩斯博士的成功能激励其他人追随他的脚步。他自己也说过,他既不是圣人也不是魔术师。Geduld导致了他的成功,这是任何一个具有毅力和决心的人都可以复制的。
参考文献
[edit | edit source]"Antonio Grillo-Lopez: Executive Profile & Biography." Bloomberg Businessweek. N.p., n.d. Web. 2012. <http://investing.businessweek.com/research/stocks/people/person.asp?personId=8006353&ticker=BIIB&previousCapId=26731&previousTitle=Deltagen%20Research%20Laboratories%20LLC>.
"Board of Directors." Onyx Pharmaceuticals. N.p., n.d. Web. 2012. <http://www.onyx.com/about-us/board-of-directors>.
Grillo-López, Antonio. "Accelerated Approval of Cancer Drugs: Improved Access to Therapeutic Breakthroughs or Early Release of Unsafe and Ineffective Drugs?." Journal of clinical oncology 27.26 (2009): 4401. Journal of Clinical Oncology. Web. 2012.
Grillo-López, Antonio. "Curing Cancer: The Rituxan Story." CHEM 92. UCSD. YORK 2622, La Jolla. 2012. Class lecture.
Mesa, Ruben. "Hodgkin's lymphoma (Hodgkin's disease)." Mayo Clinic. Mayo Foundation for Medical Education and Research, 19 Oct. 2011. Web. 2012 <http://www.mayoclinic.com/health/lymphoma/AN01209>.
"Rituxan (Rituximab) for RA, NHL, CLL, WG and MPA ." Rituxan (Rituximab) for RA, NHL, CLL, WG and MPA . Genentech, n.d. Web. 2012. <http://www.rituxan.com/index.html>.
"Rituximab Injection." PubMed Health. National Center for Biotechnology Information, 10 Mar. 2010. Web. 2012. <http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000388/>.
"The Nobel Prize in Physiology or Medicine 1908." Nobelprize.org. The Nobel Foundation, n.d. Web. 2012. <http://www.nobelprize.org/nobel_prizes/medicine/laureates/1908/>.
概述
[edit | edit source]

该化学物质的IUPAC名称是13-乙基-17-乙炔基-17-羟基-1,2,6,7,8,9,10,11,12,13,14,15,16,17-十四氢环戊并[a]菲-3-酮。它的化学式是C_21H_28O_2。左炔诺孕酮是炔诺孕酮的左旋体(D-型)。赫歇尔·史密斯在1960年代在怀斯制药公司首次合成了这种药物。这种药物主要用作紧急避孕药。
这种药物通常以片剂形式口服,更常见的名字是Plan B或事后避孕药。许多使用者错误地认为该药物是堕胎药。需要明确的是,左炔诺孕酮不会终止成熟的胎儿。该药物仅通过推迟排卵和阻碍精子迁移来阻止妊娠。左炔诺孕酮是其他避孕药中的常见成分,作用方式相同,但Plan B和其他紧急避孕药中的激素水平要高得多。
作为一种孕激素类固醇,左炔诺孕酮通过阻止促卵泡激素和黄体生成素从垂体分泌来阻止排卵。该药物还会使子宫颈粘液变得更加粘稠,这使得精子难以在子宫中游动很远,从而阻止卵子着床。
口服避孕药:低剂量的左炔诺孕酮用于单相和三相的联合口服避孕药,可用的单相剂量范围为 100-250 µg,三相剂量为 50 µg/75 µg/125 µg。在每天 30 µg 的极低剂量下,左炔诺孕酮用于一些仅含孕激素的口服避孕药。
紧急避孕:左炔诺孕酮用于紧急避孕药 (ECP),包括联合的尤氏方案(包括雌激素)和仅含左炔诺孕酮的方法。仅含左炔诺孕酮的方法使用 1.5 毫克左炔诺孕酮(一次服用或两次 0.75 毫克,间隔 12 小时),在无保护性行为后 3 天内服用,一项研究表明,在性交后最晚 120 小时(5 天)服用仍然有效。仅含左炔诺孕酮的 ECP 有许多品牌名称,包括:Escapelle、Plan B、Levonelle、Glanique、NorLevo、Postinor-2、i-pill、“Next Choice” 和 72-HOURS。左炔诺孕酮作为仅含孕激素的紧急避孕药的主要作用机制是通过抑制排卵来阻止受精。国际妇产科学会 (FIGO) 发布了一份声明: “对证据的审查表明,LNG [左炔诺孕酮] ECP 不能阻止受精卵着床。LNG ECP 产品标签中不应包含有关着床的说明。” 2012 年 6 月,《纽约时报》的一篇社论呼吁 FDA 从标签中删除有关左炔诺孕酮紧急避孕药抑制着床的无依据说法。
宫内节育器:左炔诺孕酮是米蕾娜宫内节育器的活性成分。
避孕植入剂:左炔诺孕酮是诺植入和 Jadelle 的活性成分。
Plan B 应在无保护性行为后 72 小时内服用,但在 24 小时内服用效果最佳,以防止怀孕。您可以从大多数当地药店和药房购买 Plan B,无需 18 岁及以上女性的处方。17 岁以下的任何人需要医生开具处方。Plan B 不应代替常规避孕措施,而应在以下紧急情况下使用
- 性交时没有使用避孕套,或者避孕套破裂
- 没有使用避孕措施/或者漏服了药
- 常规避孕措施失败
- 伴侣没有及时抽离
- 性行为是被强迫的。
如果已经怀孕,请不要服用 Plan B。
每个人的副作用的严重程度和发生率都不同。这些包括
- 头痛
- 眩晕
- 疲劳
- 恶心/呕吐
- 腹痛
- 乳房胀痛
- 月经不调
更严重的副作用,如瘙痒和皮疹,应立即去看医生。糖尿病患者和哺乳期妇女在使用 Plan B 时应格外小心。
在服用任何新药时,请始终注意可能存在的药物相互作用。服用 Plan B 前,请咨询医生并告知他们您目前服用的任何药物。
- http://www.parkinsons-information-exchange-network-online.com/drugdb/071.html
- http://women.webmd.com/guide/plan-b
- http://en.wikipedia.org/wiki/Plan_B_(drug)#cite_note-0
- 左炔诺孕酮 在 大英百科全书
“更好的医疗治疗并不总是需要更强的药物。化学剂的有效性取决于给药方法。因此,治疗方法通常可以通过找到最佳的药物制剂或递送系统来改善。”(马克·萨尔茨曼教授)因此,许多化学家、生物化学家、药剂师、生物工程师和化学工程师致力于改善药物的给药方法,而不是寻找更强的药物。药物给药方法分为三类:局部、非肠道和肠道。
药物给药途径是指将药物或其他化学物质插入患者或动物体内以使其被吸收到血液中并递送至目标组织的各种方法。已知有多种方法可以将药物给药到生物体内,但最简单也是最常用的方法是注射。
对于局部给药,药物直接涂抹在患处。效果局限在局部区域。局部给药的优点是药物可以由患者自行给药。不需要专业人员进行给药。通常,给药过程中不会产生疼痛。副作用很少。
药物直接涂抹在皮肤表面。这种给药方法可用于测试患者是否对某些药物或物质过敏。经皮给药还可以用于局部麻醉,以缓解切割或烧伤造成的轻微疼痛。
药物直接滴入患眼。眼药水是含有盐水的液体。眼药水的例子有冲洗眼药水、青光眼眼药水、类固醇和抗生素眼药水。
药物由患者通过嘴巴吸入。吸入给药药物可用于治疗哮喘或上呼吸道的急性感染。吸入给药的优点是身体的反应速度比口服给药快得多,因为药物不需要通过胃肠道 (GI) 道。此外,由于药物不需要通过肝脏,因此对肝脏的损害降至最低。
鼻腔给药也称为鼻喷雾。鼻喷雾的例子有鼻腔减充血剂鼻喷雾或缓解过敏的鼻喷雾。
非肠道药物递送对人体有全身性作用,这意味着药物不会专门停留在一个区域,而是作用于整个身体。在非肠道药物递送中,物质通过胃肠道 (GI) 道以外的途径给药,包括静脉注射、皮下注射、动脉内注射等。
物质被注射到静脉中。物质可以是药物。第一个静脉注射是在 1667 年给患者进行的。静脉注射的优点是反应非常迅速,药物剂量可以轻松控制,并且静脉对较高浓度的刺激性药物不敏感。然而,静脉注射也有许多缺点。首先,并不总是容易找到合适的静脉。当患者处于高压或紧张状态时,静脉会消失。其次,静脉注射可能是有毒的,因为它会引起身体的快速反应。第三,需要专业人员,患者无法自行给药。最后,准备针头消毒的成本很高。
药物被注射到肌肉中。药物可以是疫苗或抗生素。肌肉注射的优点是能够应用大量的药物,并能实现药物的可持续释放。肌肉可以作为吸附区室。当体内药物浓度较低时,肌肉可以缓慢地将药物释放到体内。然而,肌肉注射也有很多缺点。肌肉注射需要经过培训的人员进行;药物不能给患者使用。吸收有时不稳定,特别是对于溶解性差的药物。药物的溶剂可能比药物更快释放,导致药物在注射部位沉淀,从而使药物固定。
皮下 (SC, SQ) 途径是用途最广泛的给药途径之一,因为它可用于短期和长期治疗。在皮肤表面下方注射药物或植入装置是在上臂的松散间质组织、大腿前表面或腹部下部进行的。上背部也可以作为皮下给药的部位。当频繁注射时,通常需要轮换注射部位。皮下注射的药物最大剂量约为 2 毫升。针头一般长 3/8 到 1 英寸,规格为 24 到 27 号。
药物从皮下组织的吸收受与决定肌肉部位吸收速率相同的因素影响;然而,皮下组织的血管比肌肉组织少,因此吸收可能比肌肉注射后慢。但皮下给药后的吸收通常比口服给药后更快、更可预测。
皮下注射是指注射到皮肤下。胰岛素是皮下注射的一个很好的例子。皮下注射的优点是患者可以自己注射,因为不需要经过培训的人员,而且吸收虽然缓慢,但通常是完整的。可以通过在注射部位进行按摩或热敷来改善吸收。然而,皮下注射非常疼痛,刺激性药物会导致皮肤损伤,而且只能注射少量药物。
下面是说明不同类型注射差异的图片。
动脉内注射是指注射到动脉中。但这非常危险,因为动脉位于身体深处。
皮内注射是指注射到皮肤本身。例如,皮肤对某些过敏原的测试或纹身。
透皮贴剂利用药物通过皮肤的扩散来维持药物释放。例如,透皮贴剂。下面的图片展示了传统肌肉注射和透皮贴剂之间的区别。从图中可以看出,肌肉注射比透皮贴剂更痛苦。
许多药物可以制成片剂或胶囊。口服给药的优点是便携、方便、易于服用,而且不痛。它很便宜,因为片剂不需要灭菌,可以大量生产。然而,口服给药有时可能效率低下,因为高剂量或溶解度低的药物生物利用度低。药物在吸收过程中也会在肝脏中代谢,这被称为首过效应。此外,患者食用的食物也会改变药物的效果。药物也会损害胃肠道中的正常肠道菌群。最后,口服药物不能给昏迷的患者服用。
各种药物都以栓剂或灌肠剂的形式生产。
许多药物或营养物质可以通过胃管给昏迷患者服用。
http://www.ncbi.nlm.nih.gov/sites/entrez?db=mesh&term=Drug%20Administration%20Routes 1. 张良方. “药物给药和药物递送”. CENG 207 讲座 2. 加州大学圣地亚哥分校,拉荷亚. 2012 年 4 月 5 日. 讲座. 2. 郭伟伟. “关键概念”. 讲座 3. 加州大学圣地亚哥分校,拉荷亚. 2012 年 4 月 10 日. 讲座.
3. “药物给药:药物的给药和动力学:默克家庭诊疗手册”. 药物给药:药物的给药和动力学:默克家庭诊疗手册. 无人执笔,无出版日期. 网络. 2012 年 10 月 28 日. <http://www.merckmanuals.com/home/drugs/administration_and_kinetics_of_drugs/drug_administration.html>. 4. “透皮药物的原理和实践 - 全国健康联合会 - 为健康自由发声”. 透皮药物的原理和实践 - 全国健康联合会 - 为健康自由发声. 无人执笔,无出版日期. 网络. 2012 年 10 月 28 日. <http://www.thenhf.com/article.php?id=579>. 5. “药物水平的起伏:药代动力学入门”. TheBody.com. 无人执笔,无出版日期. 网络. 2012 年 10 月 28 日. <http://www.thebody.com/content/art13513.html>. 局部 对于局部药物给药,药物直接应用于患处。效果保留在局部区域。局部给药的优点是药物可以给患者使用。不需要经过培训的人员进行应用。通常,给药过程中不会有任何疼痛。副作用非常小。
表皮 药物直接应用于皮肤表面。这种给药方式可用于测试患者是否对某些药物或物质过敏。表皮给药也可用于局部麻醉,以缓解割伤或烧伤的轻微疼痛。眼药水 药物直接滴入患眼。眼药水是含盐水的液体。眼药水的例子有冲洗眼药水、青光眼眼药水、类固醇和抗生素眼药水。吸入 患者通过口腔吸入药物。吸入给药的药物可用于治疗哮喘或上呼吸道急性感染。吸入给药的优点是机体的反应速度比口服给药快得多,因为药物不需要通过胃肠道 (GI) 途径。此外,由于药物不需要通过肝脏,因此对肝脏的损害最小。鼻腔 鼻腔给药也被称为鼻喷雾剂。鼻喷雾剂的例子有减充血剂鼻喷雾剂或缓解过敏的鼻喷雾剂。
“皮外”一词指的是局部药物给药途径,通过该途径,药物、毒药、液体或其他物质通过直接皮肤涂抹进入人体。三种主要的给药途径包括:局部,其作用是局部的;肠道,其作用是非局部的;和肠外,其作用是全身性的。皮外用药从乳液、霜剂和软膏到粉剂、贴剂和酊剂。其他局部用药可以吸入或涂抹在皮肤以外的组织表面。过量使用局部用药会导致皮肤不良反应,例如瘙痒、炎症和发红;然而,如果使用得当,可以产生预期效果。
皮肤作为一层屏障,将体内结构与外部环境隔开,保持略微酸性的 pH 值,以防止病原体和其他外来入侵者。皮肤有几层:最外层称为表皮,然后是真皮,然后是最内层脂肪组织。药物通过皮肤给药的两个主要途径是
1) 经皮吸收:此过程概述了物质穿过角质层的扩散。扩散可以通过细胞间脂质途径或通过为极性化合物和离子设计的微观途径发生。由于表皮结构极其致密,细胞间隙很少,渗透需要穿过细胞膜。然而,最大的困难在于渗透真皮,真皮中扩散是通过物质的相互交织的通道进行的。
2) 经毛囊(旁路途径)吸收:皮脂腺和汗腺作为皮肤的附属物和药物吸收的通道,充当旁路,绕过角质层。整个机制通过通过皮脂的分配和扩散而进行,驱动力是浓度梯度。一个公式被用来描述这个过程:R = h/FDK
R = 扩散电阻,F = 分数面积,H = 厚度,D = 扩散率,K = 相对容量,
1) 过敏测试:抗原提取物作为穿刺皮肤测试应用于皮肤。这种方法测量皮肤对注射物质的反应性。这可以通过划痕试验、刺扎试验或皮内试验来进行。
2) 局部麻醉:局部麻醉通过麻木感觉并导致痛觉缺失,立即缓解应用部位。这些药物可以归类为酰胺类或氨基酯类,以注射、喷雾或软膏的形式给药。这些药物通常用于牙科或皮肤目的。
3) 透皮贴剂:透皮贴剂与肠外类别重叠,但一些也可能属于皮外类别。它们有很多用途
- 预防晕动症:东莨菪碱是美国食品药品监督管理局于 1979 年批准的第一个透皮贴剂。
- 避孕
- 激素替代疗法:缓解更年期症状。
- 戒烟
- 过敏测试
4) 关节炎疼痛:这些药物主要以乳膏的形式存在,用于立即和暂时缓解手部关节炎的感觉。它们有几种形式
- 反刺激剂
- 水杨酸盐
- 辣椒素
5) 伤口/皮疹:皮肤病可以用乳膏、乳液、凝胶、软膏或在某些情况下使用类固醇来治疗。
- “局部药物递送系统:综述。”药物递送系统:局部药物递送 |。N.p.,n.d. 网络。2012 年 10 月 29 日。<http://www.pharmainfo.net/reviews/topical-drug-delivery-systems-review>。
- “过敏和免疫学。”佛蒙特大学,n.d. 网络。2012 年 10 月 29 日。<http://www.uvm.edu/medicine/surgery/documents/AllergyandImmunology1.pdf>。
- “治疗关节炎疼痛的局部乳膏。”About.com 骨关节炎。N.p.,n.d. 网络。2012 年 10 月 29 日。<http://osteoarthritis.about.com/od/painrelief/a/topical_creams.htm>。
滴眼液是一种局部药物给药途径,将含有生理盐水的药物溶液滴入眼球表面。尽管大多数滴眼液通常会因患者反应而丢失或残留在眼球表面,但一些药物会通过角膜的粘膜以及结膜粘膜和部分泪液引流系统进入血液。滴眼液给药的主要优势是易于定期使用高浓度药物,以及无需通过肠道消化。通过滴眼液给药的药物可以是完全水溶液,也可以是悬浮液(浑浊)。
滴眼液是在眼科手术后或眼睛干涩严重时给患者的药物。滴眼液可以帮助清洁眼睛,使其看得更清楚。
1) 洗眼液
洗眼液是帮助润滑或替代眼泪的溶液。洗眼液不一定含有任何药物。这是最常见的滴眼液给药形式。
2) 抗生素和类固醇滴眼液
抗生素和类固醇滴眼液主要用于治疗眼部感染。它们还可以用于预防眼科手术后发生感染。
3) 青光眼滴眼液
青光眼滴眼液有助于将液体从眼睛中引流出来,从而降低眼压。青光眼滴眼液可能会有很大差异,有时会与许多其他药物组合使用,这些药物因患者的需求而异。
4) 抗组胺滴眼液
抗组胺滴眼液含有抗组胺药,可以抑制身体对过敏原的反应。它们通过抑制组胺释放到身体中起作用。它们不一定针对过敏原,但可以帮助患者更好地应对过敏原的存在。抗组胺滴眼液用于减轻眼睛的组胺反应。
5) 类固醇和抗生素滴眼液
这些滴眼液用于治疗眼部感染。必须按照规定的时间使用,以避免感染复发。首次使用时,药物可能会灼伤您的眼睛。
6) 青光眼滴眼液
这些滴眼液通过帮助眼液更好地引流来降低眼压。它们按活性成分分类。
与许多其他药物一样,滴眼液也被认为会产生一些副作用:喉咙痛、发烧、皮疹、瘙痒、头晕、肿胀。最常见的副作用之一是使用滴眼液后眼睛发红。这会导致眼睛疼痛或视力问题,应注意。但事实上,滴眼液的风险低于片剂药物,有时,可以通过使用后不要按压眼睛内角来预防这些风险。如果您没有正确使用,您不仅会感染一只眼睛,还会感染两只眼睛。
“通用名称:类固醇和抗生素滴眼液”
“青光眼药物及其副作用”
http://www.patient.co.uk/doctor/Prescribing-for-and-Administration-of-Drugs-to-the-Eye.htm#ref-1
http://development.aao.org/eyecare/treatment/eyedrops.cfm
吸入是一种药物给药途径,通过该途径,药物或吸入剂被吸入。吸入在医学上有效地用于多种麻醉剂。这种给药途径的起效非常快,这是因为肺部有毛细血管,使药物很容易快速进入血液。一旦药物进入血液,大约需要 5 到 8 秒才能到达大脑。
虽然吸入是药物给药的有效方法,但在医学上使用这种给药方法时,必须考虑三个因素。
1. 药物和材料不得刺激粘膜和肺部。
2. 控制剂量可能比其他给药方法更困难。
3. 必须持续给药,直到达到预期效果。[1]
- Hart,Carl。药物、社会和人类行为。第 12 版。麦格劳-希尔人文,2008 年。印刷。
鼻腔药物给药是许多药物局部和全身给药的一种有吸引力的选择。鼻粘膜 - 换句话说,鼻子 - 很容易接触。鼻腔药物给药是非侵入性的,几乎无痛,并且是给儿童服用药物最容易接受的方式。患者或医护人员可以在紧急情况下轻松进行应用。鼻腔药物给药可迅速产生治疗效果(局部或全身)。鼻腔给药可避免药物在胃肠道中的降解和肝脏的首过代谢。药物、载体和应用装置构成一个不可分割的三位一体。因此,它的选择对于成功开发有效的鼻腔产品至关重要。本文讨论了鼻腔给药的可行性和潜力。然而,在以下主题上仍有许多有争议的话题
- 预期用途(治疗考虑因素)
- 药物
- 载体
- 应用装置
参考
[edit | edit source]http://content.karger.com/ProdukteDB/Katalogteile/isbn3_8055/_96/_15/CUPDE40_04.pdf 定义 药物通过胃肠道进入体内,然后被血液吸收的给药途径。它可以分为三类。
类别
参考文献 药物给药途径 - P. Verma,IJPSR/Vol. I/期 I/2010 年 7 月至 9 月/第 54-59 页
介绍
[edit | edit source]口服药物给药是通过口腔给药,使药物进入消化道的过程。它被认为是最常用的给药途径,因为它方便且经济高效。通过这种途径进入的药物通常以固体剂型存在,以促进药物的高度稳定性,但它们也可以以液体剂型存在。不同的药物可以在不同的时间间隔口服,时间间隔可能是在进食前或进食后。
一般途径
[edit | edit source]大部分吸收过程在摄入后五到三十分钟内完成,但吸收通常要六到八小时才能完全完成。一旦药物通过胃部,药物需要从小肠进入血液。将肠壁与血液毛细血管隔开的膜由两层脂肪分子组成,因此需要物质是脂溶性的或可溶于脂肪才能通过。然而,即使成功地被血液毛细血管吸收后,物质仍然必须通过肝脏进行筛选过程,然后才能释放到全身循环中。肝脏中的酶能够分解某些药物的分子结构,从而减少最终进入血流的量。由于所有这些自然屏障的存在,口服给药的药物必须以故意提高的剂量水平摄入,以考虑到某些比例的药物将无法进入血流。
缺点
[edit | edit source]口服给药药物可能会由于胃肠道吸收的不可预测性而存在问题。[1] 摄入的食物等因素会改变肠道内的 pH 值、胃动力、排空时间和药物吸收速度。
抗哮喘药和抗炎药
[edit | edit source]
皮质醇或氢化可的松是一种称为皮质类固醇的化合物的类别。这种化合物是一种在医学上使用的抗炎药。科学家已经发现,呼吸道炎症会加重哮喘发作。因此,建议通过在药物中加入皮质类固醇和吸入支气管扩张剂来共同控制口服炎症,以治疗哮喘。然而,皮质类固醇可能会产生负面副作用。例如,这种化合物会抑制儿童的生长。因此,科学家开始使用合成皮质类固醇用于吸入。此外,科学家已经发现了一种可以作为抗炎药使用的合成类固醇。氟替卡松是一种将氢原子替换为氟原子的化合物,这将有助于激活该化合物用作抗炎药的能力。这种类固醇会与细胞核内的蛋白质结合,称为糖皮质激素受体,以防止体内炎症反应。
参考文献
[edit | edit source]- Shepherd,M. "药物给药 1:口服途径。" 护理时报,2011 年。<http://www.nursingtimes.net/nursing-practice/clinical-zones/prescribing/administration-of-drugs-1-oral-route/5033729.article>
Vollhardt,Peter。Schore,Neil。有机化学第六版。W.H. Freeman 公司。纽约。2011 年。
Purves,Dale,“认知神经科学原理”,Sinauer Associates, Inc.,2008 年
Levinthal,Charles,“药物、行为和现代社会”,Pearson Education, Inc.,2008 年 人类直肠是人体中可以轻松给药并充分吸收药物的通道。有时,将药物直肠给药比口服给药更可取,例如,在恶心和呕吐的情况下。通过直肠给药药物的缺点包括排便会中断吸收,以及患者接受度低(大多数患者不希望通过直肠给药)。直肠吸收药物的机制可能与胃肠道上部的机制没有区别,尽管生理环境(例如 pH 值、液体含量)存在很大差异,但水溶液和醇溶液的吸收可能非常快,这在快速抑制急性痉挛性发作(例如在儿童中)中已被证明具有相当大的治疗价值,但栓剂的吸收通常较慢,并且很大程度上取决于栓剂基质的性质、表面活性剂或其他添加剂的使用、活性成分的粒径等。有一些证据表明,直肠给药后,高清除率药物的肝脏首过消除部分避免了,例如,利多卡因。这可以通过直肠静脉血供来解释:上部与门静脉系统相连,而下部直接与体循环相连。回顾了直肠给药各种药物类别的代表物的血浆浓度数据:抗惊厥药、非麻醉性镇痛药和非甾体抗炎药、催眠镇静药和麻醉药、强镇痛药、茶碱及其衍生物、皮质类固醇、抗菌药、噻嗪铵、异丙嗪、丁溴东莨菪碱、链激酶、孕酮、麦角胺酒石酸盐和左旋多巴。只有在少数情况下,才充分证明直肠给药途径可以获得与口服途径相当的血浆浓度。直肠途径可能提供了与口服途径相同的可能性,但制剂的影响似乎至关重要。也有可能,将来具有零级释放特性的新型药物递送系统将用于直肠给药。用 2 毫升渗透泵给药茶碱已经获得了令人感兴趣的初步结果。
参考
[edit | edit source]http://www.enotes.com/rectal-medication-administration-reference/rectal-medication-administration 口服药物是通过直接将药物置于舌下进行给药的。药物迅速溶解并通过口腔粘膜吸收,进入血液循环。药物以小巧、速溶片剂、喷雾剂、锭剂或液体悬浮剂的形式配制。最常见的口服药物是硝酸甘油片剂,这是一种止痛药,在口服给药时吸收最快。口服给药方法并不总是适用。如果牙龈或粘膜有开放性溃疡或刺激部位,则不应使用口服药物。在药物吸收之前,患者不应进食、饮水、咀嚼或吞咽。必须防止吞咽药物,因为药物在消化系统中被消化后会降低药物的有效性。口服给药方法是快速作用的,在给药后 5-10 分钟内,身体就能感受到其效果。
参考
[edit | edit source]定义
[edit | edit source]肠胃外给药是指任何非口服的将药物送入体内的途径。肌肉注射、皮下注射和静脉注射是最常用的药物给药方式。
优点
[edit | edit source]肠胃外给药的一些优势在于,它可以用于对身体吸收不良或口服无效的药物。
缺点
[edit | edit source]这种药物给药方法由于所需设备数量较多,患者的疼痛感更强,因此往往比较昂贵。因此,建议由合格的医务人员进行操作。肠胃外给药的其他风险包括,在给药过程中出现的任何错误都可能导致身体受到物理伤害,或药物在体内吸收不佳。
参考文献
[edit | edit source]Martin, Shepherd. "药物给药 3: 肠胃外给药" (2011): n. page. Print.
定义
[edit | edit source]静脉注射 (IV) 是一种药物输送方法,药物直接注入血液。通过这种方法,由于药物直接进入血液,因此作用开始时间要快得多。在使用刺激性物质时,静脉输送药物可能是有益的,因为血管壁对刺激物的敏感性很低。静脉注射可以比其他方法更快地输送高浓度和剂量的药物,这可能是有益的,但也同时存在危险。
使用静脉注射的缺点
[edit | edit source]虽然静脉注射速度很快,但使用这种方法也有一些缺点。使用静脉注射的一个主要缺点是,在同一部位重复注射会导致静脉注射部位周围的区域失去强度和弹性。在最坏的情况下,如果一个特定的注射部位被刺穿多次,静脉壁就会塌陷,血液将不再流经该部位。使用静脉注射的另一个问题是感染的传播。污染的针头和注射器不应使用超过一次;也不应该在患者之间共用。[1]
参考文献
[edit | edit source]- Hart,Carl。药物、社会和人类行为。第 12 版。麦格劳-希尔人文,2008 年。印刷。
定义
[edit | edit source]肌肉注射 (IM) 是一种药物给药途径,药物被注射到肌肉中。在吸收方面,注射方法中药物到达血液的速度从快到慢依次是静脉注射、肌肉注射、皮下注射。肌肉注射因其快速起效和持久作用而备受青睐。这些注射剂注入深层肌肉组织,药物沉积后逐渐被血液吸收。
吸收
[edit | edit source]肌肉注射的主要部位有三个:手臂的三角肌、臀部和大腿。注射到手臂的吸收速度最快,而注射到臀部的吸收速度最慢。
优点
[edit | edit source]注射到肌肉中,由于血液供应充足,吸收速度快,因此刺激性较小。此外,如果需要注射大量药物,最好注射到肌肉部位,而不是皮下注射。
步骤
[edit | edit source]针头长度不应小于 1 英寸,也不应超过 1.5 英寸。建议每个成年人每次注射部位的药物最大体积为 5 毫升。
参考文献
[edit | edit source]- Hart,Carl。药物、社会和人类行为。第 12 版。麦格劳-希尔人文,2008 年。印刷。
"进行肌肉注射。"进行肌肉注射、皮下注射和皮内注射。Brookside Associates,n.d. Web.

定义
[edit | edit source]皮下注射 (SC、SQ、SubQ) 是一种药物给药方法,药物被注射到皮下组织,即真皮和表皮下方直接的脂肪组织层。由于皮下组织的血液流动有限,因此皮下注射通常用于需要缓慢吸收药物的情况。[4] 使用这种方法给药的药物必须可溶并且在低浓度下有效。这种方法常用的药物包括肝素、胰岛素、生长激素以及麻疹、腮腺炎、风疹和水痘疫苗。
注射部位和方法
[edit | edit source]皮下注射时,药物通常通过上臂松散的间质组织、大腿前表面、腹部下部或背部下部进行注射。注射时,应捏起皮肤,防止药物注射到肌肉中。建议对常规注射使用同一注射部位。如果可以捏起至少 2 英寸的皮肤,则以直角 90 度进行注射。否则,应以 45 度角注射。[5]
吸收率
[edit | edit source]可能会提高吸收率的因素包括热量、按摩、在注射部位联合使用血管扩张剂或透明质酸酶。肾上腺素可能会由于血液流动减少而降低吸收率。
参考文献
[edit | edit source]- ↑ 美国卫生系统药剂师协会 (2009-03-23). "羟考酮". 美国国家医学图书馆,MedlinePlus. 检索于 2009-03-27.
- ↑ 羟考酮, 2012 年 10 月 28 日
- ↑ 沃尔哈特,彼得。肖尔,尼尔。有机化学第六版。W.H.弗里曼公司。纽约。2011 年。
- ↑ 美国国立卫生研究院临床中心。 "患者教育:皮下注射."
- ↑ "如何进行皮下注射。"Drugs.com,无日期。网页。2012 年 10 月 28 日。 http://www.drugs.com/cg/how-to-give-a-subcutaneous-injection.html.
定义
[edit | edit source]动脉内是指在动脉内或对动脉结构进行的操作。
吸入优点
[edit | edit source]动脉内吸入的一个优点是它是一种更有效地控制注射剂量的吸入方法。患者能够控制他们将接收多少药物,因为他们能够滴定它。
吸入缺点
[edit | edit source]动脉内吸入的一个缺点是这种操作需要更长的时间。这条路线比较慢,因为肺部需要吸入它。从肺部,它必须进入全身循环。
注射优点
[edit | edit source]动脉内注射的优点是速度很快,静脉注射大约需要 15 到 30 秒,肌肉注射和皮下注射大约需要 3 到 5 分钟。另一个优点是它的生物利用度为 100%。第三个优点是单次注射可以持续数天或数月。这种方法适用于不能通过消化系统吸收的药物。如果药物过于刺激,这种方法也很适合。另一个优点是可以持续给药。
注射缺点
[edit | edit source]动脉内注射的缺点是,如果注射滥用药物,会增加成瘾和过量的风险。这是由于药物的作用速度很快。另一个缺点是患者通常无法自行注射。害怕针头和注射本身是另一个缺点,称为针头恐惧症。共用针头会导致艾滋病毒等传染病。这种给药方式是最危险的给药方式之一。动脉内注射绕过了我们体内的大部分天然防御系统。这会使患者面临健康问题。包括感染、脓肿、肝炎以及污染物或未溶解的颗粒。如果操作不当,可能会出现气泡。
定义
[edit | edit source]皮内 (ID) 给药是指将少量液体注射到皮肤的真皮层中。最常用于这种技术的是结核菌素注射器,配有 26 号针头,长度约为四分之一到二分之一英寸。注射后皮肤出现轻微肿胀是正常的。
参考文献
[edit | edit source]. "进行皮内注射。"进行肌肉内、皮下和皮内注射。Brookside Associates,无日期。网页。
介绍
[edit | edit source]经皮是指药物通过完整的皮肤扩散以达到全身分布(与局部分布相反)的过程。为了实现这种给药途径,可以使用透皮贴剂、透皮凝胶和透皮植入物等工具用于医疗目的。
途径
[edit | edit source]跨细胞途径
[edit | edit source]跨细胞途径是药物穿过皮肤的更直接的途径。在这种途径中,药物通过构成皮肤顶层(称为角质层)的死亡角质形成细胞(表皮中主要的细胞类型)的膜被运输穿过皮肤。尽管这种方法的路径距离最短,但由于药物必须穿过磷脂膜,因此会遇到阻力。
细胞间途径
[edit | edit source]细胞间途径是药物通过皮肤的更常见途径,它通过皮肤细胞之间存在的微小空间进行导航。
缺点
[edit | edit source]尽管皮肤易于接触以进行药物递送,但其防止外来物质进入人体的主要功能会限制通过这种给药途径摄入的药物类型和剂量。为了到达真皮的微循环,药物必须设法穿过表皮和真皮,这两个是皮肤的两个重要层。
参考文献
[edit | edit source]- McCarley, K.D & Bunge, A.L. (2001)。 "经皮吸收药代动力学模型综述。"J 药学科学。90: 1699–1719。
- Hadgraft, J. (2001)。 "皮肤屏障功能的调节。"皮肤药理学应用皮肤生理学。14(1): 72-81。
介绍
[edit | edit source]透粘膜是指药物通过粘膜扩散的给药途径。这可以指吸入、鼻腔、舌下、阴道、直肠或眼部途径。
吹入
[edit | edit source]吹入是指吸入物质的行为。它通常用作治疗鼻窦和肺部疾病的呼吸道药物的途径。这种给药途径通常用于精神活性药物,因为它能够更快地扩散进入血液,并且能够使药物绕过血脑屏障。对于这种方法,生物利用度高于口服途径的生物利用度。可卡因是一种常见的利用吹入途径的药物。
舌下给药
[edit | edit source]舌下给药是指利用舌头下面的组织将药物扩散到血液中的过程。专为这种途径设计的药物包括巴比妥类药物、酶类、类固醇和心血管药物。与其他跨粘膜途径一样,舌下给药通过舌头下面的粘膜扩散。此过程比口服途径更直接,但也更难执行,因此降低了药物在进入血液之前因唾液酶降解的风险。
- 威廉·H·弗雷。“绕过血脑屏障,将治疗剂递送到大脑和脊髓。”药物递送技术。
- mendelson JE, Coyle JR, Lopez JC, Baggott MJ, Flower K, Everhart ET, Munro TA, Galloway GP, Cohen BM。“舌下服用天然κ-阿片类药物沙尔维辛A对人体的缺乏影响:安慰剂对照试验”。舌下研究,www.maps.org。精神药理学(柏林)。检索于 2012 年 1 月 26 日。
吸入是一种药物给药途径,通过该途径,药物或吸入剂被吸入。吸入在医学上有效地用于多种麻醉剂。这种给药途径的起效非常快,这是因为肺部有毛细血管,使药物很容易快速进入血液。一旦药物进入血液,大约需要 5 到 8 秒才能到达大脑。
虽然吸入是药物给药的有效方法,但在医学上使用这种给药方法时,必须考虑三个因素。
1. 药物和材料不得刺激粘膜和肺部。
2. 控制剂量可能比其他给药方法更困难。
3. 必须持续给药,直到达到预期效果。[1]
- Hart,Carl。药物、社会和人类行为。第 12 版。麦格劳-希尔人文,2008 年。印刷。
药代动力学,缩写为“PK”(源于古希腊语pharmakon“药物”和kinetikos“与运动有关”),是药理学的一个分支,专注于生物系统对化学物质的影响。它涉及药物在我们体内的三个主要阶段,即吸收、分布和排泄。该领域主要适用于化学药物,但也适用于被生物体摄入或外部输送的物质,例如营养物质、代谢产物、激素、毒素等。
药代动力学通常与药效动力学一起研究。两者不应混淆;药代动力学描述为身体对药物的作用,而药效动力学描述为药物对身体的作用。药代动力学扩展到药物吸收和分布的机制,药物效应开始的速率以及效应的持续时间,药物在体内的化学转化(最有可能由酶介导),以及药物的排泄效应和途径。
所有药物,无论以何种方式递送,在考虑其随时间推移的影响时,都有一些共同特征。最初有一个间隔,即潜伏期,在此期间药物的浓度在血液中升高,但尚未高到足以检测到药物效应。此潜伏期持续多长时间,通常与药物的吸收时间有关。随着药物浓度的继续升高,效应将变得更强。
一种药物的耐受效应可能自动诱导另一种药物的耐受。这种效应称为交叉耐受,通常见于酒精、巴比妥类药物和一类抗焦虑药物的生理和心理效应。由于交叉耐受,酒精中毒者通常会对巴比妥类药物产生耐受性,这在接受麻醉手术时可能是危险因素。另一方面,如果我们可以通过服用另一种药物来缓解一种药物的戒断症状,那么这两种药物就表现出交叉依赖性。实际上,一种药物可以替代另一种已停用药物产生的任何生理效应。
药代动力学及其在药物剂量中的作用:药代动力学是支持应用治疗学的主要系统性调节。它是测试药物在体内的持续时间及其对身体的影响的数学基础。当患者需要药物时,医生会开出针对特定疾病的适当药物和剂量的处方。此剂量在药物使用过程(DUP)中进行监测。医生和药剂师始终确保患者没有遭受与药物相关的疾病。一旦确认了此说法,就可以进行临床诊断,药剂师可以应用 DUP 来保证剂量和程序适合患者。方案是根据患者处理药物的能力制定的。药物在体内有四个阶段:吸收、分布、代谢和排泄。药物浓度是根据这一基础制定的。当患者了解这些阶段时,药物就可以分配给患者。药剂师必须确认药物、剂量和方案是否合适,并且患者能清楚地理解和接受。临床药代动力学是药剂师必须熟悉和掌握的关键基础,也是药剂师成功进行药学护理所需的素质。理想情况下,药物的强度是在激活部位(受体)处计算的。但是,由于这不可能,因此在血液、唾液、尿液和/或脑脊液中测量药物水平。反应速率:为了准确地应用 ADME 方法,必须考虑这些步骤的速率。反应速率,反应持续的速度,可以是零级或一级。分布容积:分布容积不是一个实际的定量体积值,而更像一个表观体积。它是一种测量药物浓度的指标。它是在身体中溶解药物所需的等量血浆的体积。由于身体不是一个同质实体,因此很难精确测量浓度。药物在身体的各个部位的浓度可能不同。但是,重要的是要注意,浓度在整个身体内将成比例,并且可以相应地合理化这些值。公式 Vd= X/Cp 用于将药物量转换为其浓度。Vd 是分布容积,X 是组织中的药物量,Cp 是身体特定部位中的药物量。使用该公式,如果药物的分布容积很大,并且与准确的体积读数不匹配,则认为药物高度分散在组织中。如果药物的分布容积与准确的体积读数相匹配,则认为药物分散不成功,并且严格地包含在血浆中。药物清除率:药物清除率 (CL) 是通过代谢和排泄功能,血管部分每单位时间内无药物的血浆体积。如果确认一种特定药物从一级动力学中清除,那么其清除率在整个过程中保持不变。CL=k X Vd,其中 k 是一级消除速率常数,Vd 是分布容积。多次用药:许多患者需要服用多次药物才能有效。当药物被身体消耗时,药物会在体内积聚,浓度会升高,直到达到稳态。稳态发生在消耗的药物浓度等于相同时间段内消除的药物浓度时。在稳态下,药物的血浆浓度以及高点和低点在整个过程中保持恒定。达到稳态所需的时间取决于药物的半衰期。剂量越大,稳态水平就越高。当给药间隔小于半衰期值时,积累越大,稳态水平越高。在给药间隔与药物的半衰期相比大得多的情况下,将不会有积累。许多药物的给药间隔与药物的半衰期成正比,但与推荐剂量或达到稳态所需的时间无关。
隔室模型是药物在体内分散的药代动力学中引入的一种数学表示。它是对药物在体内引入后药代动力学过程的模拟。科学家通常研究一室或二室模型[1]。
单室模型是一个封闭的均匀系统,其中给药的药物在一个身体器官的单一单元中释放和扩散,没有吸收,这是一个理想条件[1]。单室模型遵循这样一个理念,即一旦药物进入体内,它就会立即在整个身体中均匀分布,达到平衡。这是因为该模型将身体描绘成一个动力学均匀的单元。然后将血浆中药物的浓度(也定量地表示组织的变化)线性绘制,从而表示一个单室模型。单室模型的一个例子是静脉注射,没有吸收。该模型的动力学特征由药物总量 (M)、药物浓度 (C)、体液量 (V)、一级消除常数 (k) 和扩散时间 (t) 决定。[2]
三个变量的含义如下:
M: total mass of drug
V:体液量
C:药物浓度
单室模型的质量平衡
dM/dt=-kM
M=M_0 e^(-kt)
M=CV
C=M_0/V e^(-kt) ,when M_0/V=C_0
变量的含义如下:
k:一级消除常数
C0 : 给药药物的初始浓度
t:药物扩散时间
C/C0:关于时间的速率常数
当 t = 0 时,我们可以得到药物的最大浓度,也就是初始浓度:Cmax = C0
由此,我们也可以知道:Mmax = M0
单室模型中的循环半衰期 (t1/2) 计算
lnC=ln M_0/V-kt
lnC_0=ln M_0/V
ln C_0/2=ln M_0/V-kt_(1/2)
因此,
t_(1/2)=ln2/k
具有吸收的单室模型
[edit | edit source]单室模型,具有吸收,是一个封闭系统,其中给药的药物从透皮贴剂扩散到血流中。
变量与图 1-1 相同,除了:
D:初始药物吸收室的总量
ka:吸收常数
具有吸收的单室模型的质量平衡
dM/dt = kaD-kM
dD/dt = -kaD
D=D0 在 t=0 时
因此,
D=D0e-kat ⇒ dM/dt=kaD0e-kat-kM, M=0 at t=0
最后,M =D_0 k_a/(k-k_a )(e^(-k_a t)-e^(-kt))
该模型可以与治疗窗口相关联
双室模型
[edit | edit source]双室模型将身体分为两个隔室:中央隔室和外周隔室。中央隔室包括血液和血流丰富的器官,例如肝脏、肾脏、心脏、大脑等。外周隔室包括血流较差的组织,例如肌肉、瘦肉组织、脂肪等。该模型遵循这样一个理念,即一旦药物进入体内,它就会在中央隔室和外周隔室之间分配。但是,两个隔室之间不会达到平衡。
ADME
[edit | edit source]药代动力学是量化药物在体内系统中时间进程及其相应作用的基础。药物在体内时间跨度的四个过程可以描述如下:
- Absorption - 药物进入体内的过程
- Distribution - 药物分散到血流和组织中的过程
- Metabolism - 母体化合物转化为子代代谢物的过程
- Excretion - 药物从体内消除的过程
这些过程通常被称为 ADME,负责药物在药物给药过程中不同时间点在血流中的不同浓度。通常,药物的有效性取决于其在体内的浓度。给药部位和剂量等其他因素也会影响药物的药代动力学特性。
或者,可以使用 LADME 来代替 ADME。L 在 ADME 方案中添加了 Liberation 过程,即药物从其载体(通常是其保护涂层或类似材料)中释放的过程。
科学家如何研究药代动力学?
[edit | edit source]由于药代动力学领域的科学家正在追踪药物在体内的作用,因此完美的时机对于研究人员来说是一个重要的要求。科学家必须确定药物应该在何时何地靶向体内。但是,追踪药物并不容易。即使科学家已经了解药物如何通过身体的策略,但他们无法实际看到药物的去向。因此,科学家在研究中使用数学和化学工具。
数学工具:数学提供了模型和精确的方法来测量体液,帮助研究人员确定他们感兴趣的目标。血液和尿液的测量可以回答以下问题:药物在哪里,以及药物在给定时间内分解了多少。同时,肝酶的血清水平可以帮助预测将吸收多少药物。
化学的应用:药物与生物体之间的相互作用实际上是药物分子与生物体内部分子之间的一系列化学反应。因此,了解药物在生物环境中的反应方式对于预测身体所需的药物量或身体能够承受的最大药物量是必要的。
使用药代动力学的进展
[edit | edit source]通过使用可卡因药代动力学模型,科学家已经看到了使用酶疗法治疗药物滥用的可喜效果。研究可卡因代谢酶,观察它是否可以阻止药物进入大脑并导致产生生理效应,可能会导致抗可卡因药物生产的进步。该模型可以很容易地转化为其他具有生理作用的药物的治疗方法。 [1]
动力学
[edit | edit source]为了正确理解 ADME 的药代动力学过程,必须检查这些过程的速率。每个过程进行的速率或速度遵循零级或一级动力学。
零级动力学
在零级反应中,反应速率与反应物浓度无关;速率是恒定的。零级反应的速率定律为
- r = k
例如,如果药物A以恒定速率从体内排出,根据零级动力学,排泄速率可以描述为
- dA/dt = k
一级动力学
在一级反应中,反应速率仅取决于一个反应物的浓度,即使存在多个反应物也是如此。一级反应的速率定律为
- -r = k[A]
其中 [A] 是反应物 A 的浓度。如果相同的药物 A 通过一级动力学从体内排出,速率定律将写成
- dA/dt = -k[A]
大多数药物通过一级动力学进行,生物系统中的 ADME 过程通常也遵循一级动力学。
参考文献
[edit | edit source]美国卫生与公众服务部 - 药物设计
Zheng F, Zhan C-G (2012) 人体可卡因药代动力学的建模揭示了开发针对药物滥用酶疗法的可行性。PLoS Comput Biol 8(7): e1002610. doi:10.1371/journal.pcbi.1002610
Levinthal, Charles, "Drugs, Behavior, and Modern Society", Pearson Education, Inc., 2008
PTP1B
[edit | edit source]PTP1B(蛋白酪氨酸磷酸酶 1B)是一种非跨膜酶,存在于内质网 (ER) 上。它的重要性在于它是胰岛素和瘦素信号传导的负向调节剂。PTP1B 去磷酸化,或者换句话说,从胰岛素受体 IR 和其主要底物(称为胰岛素受体底物蛋白 (IRS 蛋白))中去除磷酸基团。在瘦素中,PTP1B 从 Janus 激酶 2(称为 JAK2)中去除酪氨酸激酶的磷酸基团。最近,它被发现是肿瘤发生的一个促成因素,并且与乳腺癌的直接联系更为密切。它也被认为是一个潜在的药物靶点,因为抑制它可能导致 2 型糖尿病、肥胖症和某些形式的癌症的停止。
PTP1B 的结构
[edit | edit source]PTP1B 的结构由大约 800 个残基组成。该蛋白质由一个 N 端催化磷酸酶结构域组成,后面跟着一个调节区和一个膜定位结构域。它连接到内质网。
PTP1B 最初被发现时,被发现可以抑制胰岛素和瘦素受体。PTP1B 通过去磷酸化胰岛素受体 (IR) 及其主要底物(如 IRS 蛋白)来实现这一点。反过来,这会导致患者患上糖尿病。Ptp1b 基因敲除小鼠的实验表明,抑制这种酶可以让人们保持苗条和精力充沛,而无论他们吃什么或吃多少。当这种酶在小鼠体内失活时,它们对胰岛素变得高度敏感,即使在高脂肪饮食下也能保持苗条。酶失活的特定组织部位似乎并不重要,所有实验都指向了相同的结果:健康且精力充沛的小鼠,胰岛素敏感性提高,葡萄糖耐受性增强。
PTP1B 是一种已知表达非常丰富的酶。它由一个 N 端催化磷酸酶区域组成,长度在 1 到 300 个残基之间,一个调节区域,长度在 80 到 100 个残基之间,最后是一个膜定位区域,长度在 400 到 435 个残基之间。膜定位区域将 PTP1B 酶连接或结合到内质网的胞质面。PTP1B 的表达及其催化活性通常受到四种机制的严格控制,这些机制有时会协同作用:氧化、磷酸化、SUMO 化和蛋白水解。
PTP1B 可以通过可逆和不可逆氧化在体内进行调控。Cys 215 是其活性位点之一的氨基酸,位于一个异常酸性的环境中,因此在生理 pH 值下会发生去质子化。这将氨基酸转化为催化过程中极好的亲核试剂。一旦发生这种转化,Cys 215 就会变得容易受到其他含有氧气的高反应性物质的失活作用。根据用于修饰 PTP1b 的含有氧气的高反应性物质的不同,酶会被氧化成不同的氧化状态。
通过晶体学分析,结果表明,如果使用过氧化氢,PTP1B 的亚磺酸形式会通过氧化过程转化为失活的环状亚磺酰胺状态。随着该过程的进行,活性位点也会发生构象变化,这导致隐藏的酪氨酸氨基酸(位于磷酸酪氨酸结合环中)暴露出来。预测该过程是可逆的。相反,如果酶被氧化到亚磺酸或磺酸状态,该过程将是不可逆的。
虽然丝氨酸和酪氨酸在多个位置发生磷酸化,但磷酸化的影响仍然存在争议。对酶上不同丝氨酸残基磷酸化的研究产生了相互矛盾的结果。例如,蛋白激酶 C 在中期或响应外部刺激(如渗透压)对 S378 和 S352 的磷酸化不会显着改变酶的活性水平。然而,当 S50 被 AKT 磷酸化时,酶去磷酸化胰岛素受体的能力下降。有趣的是,当相同的 S50 残基被 CDC 样激酶 1 和 2 磷酸化时,酶的磷酸酶活性提高了 2 倍。同样,对酶上酪氨酸位点 (Y66、Y152、Y153) 磷酸化的研究表明,这些对 PTP1b 的改变可以增加或降低蛋白质的活性。
最近发现,小型泛素相关修饰蛋白 (SUMO) 对许多细胞功能起着重要的调节作用。SUMO 共轭显着调节许多蛋白质特征,例如稳定性、定位、相互作用和活性。发现 PTP1B 与 SUMO E3 连接酶相互作用,从而促进 SUMO 对 PTP1B 的修饰。酶活性降低,因此 PTP1B 对底物的活性降低。SUMO 修饰发生的具体位置尚不清楚。然而,人们观察到 PTP1B 在位于核周区域和 C 末端的点状结构中积累。重要的是要注意,PTP1B 的 ER 靶向域的存在对于最大限度地发生 SUMO 化是必要的。
钙蛋白酶是一种蛋白酶,它介导 PTP1B 的 ER 靶向部分 (C 端) 的切割。这发生在被激活的血小板中,结果是酶被激活。研究表明,当小鼠体内的钙蛋白酶-1 被破坏时,蛋白质酪氨酸磷酸化水平会降低。对这些小鼠血小板的分析表明,PTP1B 的数量大幅增加。这表明,当 C 端被切割时,PTP1B 会被切割成无活性片段(片段)。支持蛋白水解(即蛋白质 PTP1B 分解成无活性片段)的证据是与钙蛋白酶-1 丢失相关的酪氨酸磷酸化缺陷在 Capn1 中得到恢复,以及血小板的聚集。

重要的是要注意,其他报告表明,当 PTP1B 可逆地被氧化时,它会被钙蛋白酶介导的催化域切割而失活。因此,有些人认为,酶是通过 C 端切割而被激活还是通过完全蛋白水解而失活(这两个过程都是由钙蛋白酶介导的)实际上取决于 PTP1B 的氧化状态。然而,迄今为止还没有关于除血小板以外的任何其他细胞类型中钙蛋白酶导致酶失活的报道。
由于 PTP1B 位于 ER 的胞质面,因此这种酶如何遇到并去磷酸化其许多不同的底物的问题被提了出来。迄今为止,提出了四种可能的机制。首先,如果底物是膜结合受体蛋白酪氨酸激酶(如胰岛素受体),则通过囊泡介导的内吞作用过程,这些被激活的受体会被内化并与 PTP1B 酶接触。其次,来自基于生物发光共振能量转移和基于荧光共振能量转移的活体图像的证据支持 PTP1B 可能在胰岛素受体生物合成过程中对其进行去磷酸化。第三,PTP1B 结合的 ER 部分是可拉伸的。这使得 PTP1B 与质膜上的底物之间的相互作用成为可能。最后,衔接蛋白将 PTP1B 连接到其底物,通过形成三元复合物促进它们的相互作用。除了这些机制之外,该酶还具有结合基序,这些基序促进其与底物(如 IR)的相互作用。研究表明,这些基序之一是位于催化域的 β9-β10 转折处的 Y152,它允许 PTP1B 与 IR 二聚体的背面相互作用。
Ptp1b 基因敲除小鼠的实验表明,抑制这种酶可以让人们保持苗条和精力充沛,而无论他们吃什么或吃多少。当这种酶在小鼠体内失活时,它们对胰岛素变得高度敏感,即使在高脂肪饮食下也能保持苗条。酶失活的特定组织部位似乎并不重要,所有实验都指向了相同的结果:健康且精力充沛的小鼠,胰岛素敏感性提高,葡萄糖耐受性增强。
到目前为止,尚未发现 PTP1b 是一种肿瘤抑制基因,但有证据表明它可能是一种细胞生长的负调节因子。该酶可以促进细胞死亡(凋亡),因此可能是一种肿瘤抑制基因。抑制这种酶似乎可以非常简单地解决许多健康问题,有些人可能想知道为什么没有更快地采取行动,但正如我们所知,生物学并非如此简单。PTP1B 抑制的总体影响尚不清楚,一些研究已经表明,抑制这种酶会促进某些类型的癌症。一些人类癌症(例如乳腺癌和卵巢癌)显示出 PTP1B 的升高水平。对于乳腺癌,ErbB2 的激活会导致 PTP1B 表达水平升高。使用转基因小鼠进行的研究表明,PTP1B 是 ErbB2 诱导的乳腺肿瘤发生的正调节因子。没有 PTP1B 的小鼠在 ErbB2 发病时间上延迟很大,而过表达 PTP1B 的小鼠则会发生乳腺肿瘤。Julien 等人在小鼠癌症模型中使用 PTP1B 抑制剂,结果表明这种抑制剂有效地保护了小鼠免受 ErbB2 致癌基因的影响。然而,即使在 PTP1B 缺陷模型中,由多瘤病毒中 T 抗原引起的乳腺肿瘤仍然可以发生。因此,PTP1B 被认为是一种在致癌信号传导中起选择性但贡献作用的酶。
1. Yip, Shu-Chin, Sayanti Saha, and Jonathan Chernoff. "PTP1B: A Double Agent in Metabolism and Oncogenesis." Trends in Biochemical Sciences 35.8 (2010): 442-49. Print.
2. "PTP1B Research for Dummies." PTP1B Research for Dummies and Why You Need To Know It | GoGetThin™. N.p., n.d. Web. 20 Nov. 2012. <http://blog.gogetthin.com/leptin-and-weight-loss/ptp1b-smart-weight-loss>.
3. “PTP1B”。维基百科。维基媒体基金会,2012 年 6 月 17 日。网页。2012 年 11 月 20 日。<http://en.wikipedia.org/wiki/PTP1B>。
4.维基百科贡献者。“钙蛋白”。维基百科,自由的百科全书。维基百科,自由的百科全书,2012 年 8 月 20 日。网页。2012 年 11 月 22 日。
苯丙胺是一种食欲抑制剂,不溶于水。服用苯丙胺后,神经和大脑会受到刺激,导致心跳加快、血压升高,最终导致食欲下降。这种药物以治疗嗜睡症和注意力缺陷多动障碍 (ADHD) 而闻名。嗜睡症是一种睡眠障碍,其特征是过度睡眠。ADHD 的特征是注意力不集中、过度活跃或两者兼而有之。

孕妇和母乳喂养的母亲禁止服用苯丙胺,因为可能会出现意外的药物影响,从母亲传递给婴儿,除非他们在事先咨询过医生。以下特征的人不应该服用苯丙胺
- 心脏问题
- 高血压
- 动脉粥样硬化(动脉硬化,由动脉中积累的脂肪引起)
- 甲状腺机能亢进(甲状腺中甲状腺激素过量)
- 青光眼(眼部神经损伤)
服用不足的情况下,如果已经是晚上,请在下次预定时间服用苯丙胺,因为如果在深夜服用,苯丙胺可能会导致失眠。服用过量的情况下,应立即寻求医生的帮助。过量服用苯丙胺的后果很严重;包括幻觉、恶心、腹泻、癫痫发作等等。
以下是苯丙胺的常见副作用;但是,应将这些副作用告知医生。如果出现严重副作用,则停止服用苯丙胺。如果出现轻微副作用,则继续服用苯丙胺。
- 严重副作用
- 过敏(尤其是在喉咙、嘴唇、脸部、舌头上)
- 心律不齐
- 高血压
- 幻觉
- 轻微副作用
- 焦虑
- 轻微头痛
- 失眠
- 腹泻/便秘
- 阳痿
如果同时服用其他药物,苯丙胺可能无法正常发挥作用,或者如果正在服用其他药物,可能需要调整剂量。以下是一些可能影响苯丙胺的药物
- 胰岛素
- 降压药
- 治疗前列腺肥大的药物
- 抗抑郁药(尤其是三环结构的抗抑郁药)
- 抗组胺药
- 治疗精神病的药物
http://www.drugs.com/amphetamine.html http://www.nlm.nih.gov
保幼激素是一种用于控制昆虫发育的化合物。因此,它有助于预防疟疾、黄热病和西尼罗河病毒等虫媒疾病。
保幼激素或 JH 由称为 Hyalophora cecropia L. 的雄性蚕蛾产生。一旦昆虫感染了 JH,它会在昆虫体内产生变态,从而阻止昆虫产卵。因此,JH 可以帮助控制疾病。然后科学家们创造了合成的和分离的天然 JH。JH 的物质随后在实验室中生产,它允许生产更稳定和生物活性更强的物质。其他合成化合物,如甲基嘧啶,也有助于预防虫媒疾病。它比 JH 的生物活性高得多,它可以攻击蚊子、跳蚤和蚂蚁。甲基嘧啶本身不会杀死昆虫,但该物质会在昆虫卵达到成虫之前杀死它们。因此,甲基嘧啶和 JH 用于防止昆虫发育和传播疾病。
沃尔哈特,彼得。肖尔,尼尔。有机化学 第 6 版。W.H.弗里曼与公司。纽约。2011 年
如果一种或多种氨基酸缺失或过量,会导致严重后果。一种称为苯丙酮尿症 (PKU) 的遗传性疾病是由人体无法清除多余的苯丙氨酸引起的。PKU 是一种常染色体隐性遗传病,这意味着只有当父母双方都携带与该病相关的基因版本时,才会患上该病。PKU 患者出生时就缺少分解苯丙氨酸氨基酸的酶。高水平的 Phe 会积累,对大脑有剧毒,因此,PKU 会导致智力障碍。然而,苯丙氨酸是一种必需氨基酸——人体不能没有它。饮食和基因都会导致 PKU,因此任何控制体内苯丙氨酸供应的方法都可以预防该病。
苯丙酮尿症 (PKU) 是遗传的,这意味着它会遗传给家族。父母双方都必须传递有缺陷的基因,婴儿才会患病。这被称为常染色体隐性性状。PKU 婴儿缺少一种叫做苯丙氨酸羟化酶的酶,这种酶是分解一种叫做苯丙氨酸的必需氨基酸所必需的。这种物质存在于含有蛋白质的食物中。没有这种酶,苯丙氨酸和两种密切相关的物质会在体内积聚。这些物质对中枢神经系统有害,会导致脑损伤。
苯丙氨酸在人体黑色素的生成中发挥作用,黑色素是负责皮肤和头发颜色的色素。因此,患有这种疾病的婴儿往往比没有这种疾病的兄弟姐妹皮肤、头发和眼睛颜色更浅。
其他症状可能包括
- 智力和社会技能发展迟缓
- 头围明显低于正常
- 活动过度
- 手臂或腿部的抽搐运动
- 智力障碍
- 癫痫发作
- 皮肤疹
- 震颤
- 手部摆放位置异常
- 如果这种疾病没有得到治疗,或者没有避免含有苯丙氨酸的食物,那么可能会在呼吸、皮肤和尿液中检测到“老鼠”或“霉味”。这种特殊的气味是由于体内苯丙氨酸物质积聚造成的。
PKU 可以通过简单的血液测试轻松检测出来。美国所有州都要求对所有新生儿进行 PKU 筛查测试,作为新生儿筛查项目的一部分。该测试通常是在婴儿离开医院之前,从婴儿身上抽取几滴血进行的。
如果最初的筛查测试呈阳性,则需要进行进一步的血液和尿液测试以确认诊断。
然而,好消息是 PKU 疾病很容易诊断,如果存在,PKU 是一种可治疗的疾病。医生通过开具终身限制性饮食来治疗 PKU 患儿。治疗包括一种极低苯丙氨酸的饮食,尤其是在孩子生长的时候。必须严格遵循饮食。这需要注册营养师或医生的密切监督,以及父母和孩子的配合。那些继续在成年后坚持饮食的人,身体和心理健康状况会更好。“终身饮食”已成为大多数专家推荐的标准。这在受孕前和整个妊娠期尤其重要。
苯丙氨酸在某些食物中含量很高,例如牛奶、鸡蛋和含有阿斯巴甜(NutraSweet)的人工甜味剂的无糖饮料是苯丙氨酸的丰富来源。这种饮食很严格,要求人们避免这些食物以及许多其他食物,例如肉类和鱼类、乳制品、面包、坚果,甚至一些蔬菜。因此,PKU 患者必须服用特殊的无苯丙氨酸维生素/矿物质补充剂,以确保他们获得这些食物中丰富的其他必需氨基酸的充足量。例如,有一种名为 Lofenalac 的特殊婴儿配方奶粉是为 PKU 婴儿制定的。它可以作为一种极低苯丙氨酸的蛋白质来源,并在生命中始终使用,并且平衡了剩余的必需氨基酸。另一个例子是服用鱼油等补充剂来代替标准无苯丙氨酸饮食中缺少的长链脂肪酸,以帮助改善神经发育,包括精细运动协调。可能还需要其他特定补充剂,例如铁或肉碱。
如果严格遵循饮食,从孩子出生后不久就开始,预后预期会非常好。如果治疗延迟或病情未得到治疗,大脑会受到损伤。学校学习能力可能会轻微受损。如果不能避免含有苯丙氨酸的蛋白质,PKU 会导致生命第一年结束时出现智力障碍。如果疾病未得到治疗,会导致严重智力障碍。多动症 (ADHD) 似乎是那些不坚持极低苯丙氨酸饮食的人中最常见的问题。
酶测定可以确定父母是否携带 PKU 基因。绒毛膜绒毛取样可以在妊娠期间进行,以筛查未出生婴儿是否有 PKU。
对于患有 PKU 的女性,在怀孕之前和整个怀孕期间严格遵循低苯丙氨酸饮食非常重要,因为这种物质的积累会损害发育中的胎儿,即使孩子没有遗传缺陷基因也是如此。
- ↑ 郑 F,湛 C-G(2012)人类可卡因药代动力学模型揭示了开发药物滥用酶疗法的可行性。PLoS Comput Biol 8(7):e1002610。doi:10.1371/journal.pcbi.1002610
- ↑ 美国卫生与公众服务部。健康化学。2006 年 10 月。<http://www.nigms.nih.gov>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- ↑ 美国卫生与公众服务部。健康化学。<http://www.nigms.nih.gov>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- ↑ 苯丙酮尿症。“<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
- 美国卫生与公众服务部。新遗传学。2006 年 10 月。<http://www.nigms.nih.gov>。
- "苯丙酮尿症。“国家卫生研究院。<http://www.nlm.nih.gov/medlineplus/ency/article/001166.htm>。
1928 年,科学家亚历山大·弗莱明爵士发现了一种抗生素青霉素,用于破坏细菌菌落。青霉素的结构是 C6H5CH2,并含有 β-内酰胺环,可激活抗生素对抗细菌的攻击。然而,一些细菌无法被抗生素破坏,因为它们有一种酶青霉素酶,可以抵抗 β-内酰胺环。另一种名为泰格西林的抗生素可以克服细菌的酶,从而允许抗生素攻击细菌。因此,这种抗生素可以抵抗皮肤和内脏的感染。因此,每年都会产生新的抗生素来帮助消除感染。
Vollhardt,Peter。Schore,Neil。有机化学第 6 版。W.H. Freeman and Company。纽约。2011


呋塞米是一种利尿剂。服用这种药物的患者可能会出现排尿频率增加和尿量增加。因此,患者可能会出现便秘和血清钠水平升高;以及排泄过量的钾、钙、磷和镁。
服用这种药物的患者应减少钠的摄入量,以避免液体潴留,并且可能需要增加钾、磷、钙和镁的摄入量。
1. 增加钾摄入量的饮食
对于谷物,推荐的食物是麸皮麦片或松饼,以及格兰诺拉麦片。
对于蔬菜,推荐的食物是朝鲜蓟、西兰花、抱子甘蓝、绿叶蔬菜、羽衣甘蓝、苤蓝、防风草、芜菁甘蓝、蘑菇、白薯和红薯、菠菜、甜菜、冬南瓜、新鲜或罐装番茄、番茄汁或蔬菜汁、西葫芦。
对于水果,推荐的食物是杏、鳄梨、香蕉、哈密瓜、枣、无花果、葡萄柚汁、奇异果、橙汁、瓜类、芒果、油桃、木瓜、梨、石榴、李子和李子汁、葡萄干。
对于牛奶,推荐的食物是脱脂、低脂、全脂、巧克力和酪乳、原味或含水果的酸奶、豆奶。
对于肉类和豆类,推荐的食物是罐装、干、新鲜或冷冻的豆类和扁豆、烤牛肉或碎牛肉、鸡肉、蛤蜊、蟹肉和新鲜、冷冻或罐装鱼类、坚果和坚果酱、猪肉和火鸡、大豆。
其他,推荐的食物是巧克力、糖蜜、薯片、麦芽。
对于上面提到的食物,没有要避免的食物,这些食物可以增加钾。
2. 增加钙摄入量的饮食
对于谷物,推荐的食物是所有食物。
对于蔬菜,推荐的食物是所有食物,尤其是菠菜和秋葵。
对于水果,推荐的食物是所有食物。
对于牛奶,推荐的食物是牛奶、酸奶、奶酪。
对于肉类和豆类,推荐的食物是所有食物,尤其是大豆。
对于上面提到的食物,没有要避免的食物,这些食物可以增加钙。
3. 增加磷摄入量的饮食
对于谷物,推荐的食物是所有食物,尤其是燕麦片和麦芽。
对于蔬菜,推荐的食物是所有食物,尤其是带皮烤土豆。
对于水果,推荐的食物是所有食物。
对于牛奶,推荐的食物是所有食物,尤其是奶酪、脱脂、低脂或普通牛奶。
对于肉类和豆类,推荐的食物是所有食物,尤其是肉类、干豆类、坚果、大豆和豆腐。
对于上面提到的食物,没有要避免的食物,这些食物可以增加磷。
4. 增加镁摄入量的饮食
对于谷物,推荐的食物包括麸皮麦片、麸皮松饼、燕麦片、糙米、全麦意大利面和细面条。
对于蔬菜,推荐的食物包括朝鲜蓟、鳄梨、绿叶蔬菜、秋葵、带皮烤土豆、菠菜、瑞士甜菜。
对于水果,推荐的食物尚不清楚。
对于牛奶,推荐的食物包括蛋奶酒、巧克力牛奶、豆奶。
对于肉类和豆类,推荐的食物包括干豆(黑豆、白豆、芸豆、利马豆和菜豆)和豌豆、鱼(大比目鱼、黄鳍金枪鱼)、大豆。
对于其他,推荐的食物包括坚果(杏仁、腰果、核桃、榛子、栗子、混合坚果、花生)和花生酱、南瓜籽或葵花籽、豆腐、小麦胚芽。
对于上述提到的食物,没有需要避免的食物来增加镁含量。
"营养护理手册" <http://nutritioncaremanual.org/content.cfm?ncm_content_id=93007>

维生素 C 被称为抗坏血病维生素,由科学家发现。一些人甚至称它为己糖醛酸或抗坏血酸。这种维生素可以用来治疗坏血病。对于那些不知道的人来说,坏血病是一种由缺乏维生素 C 引起的疾病;结果,它会导致过去愈合的伤口再次裂开,并导致牙龈肿胀甚至出血。坏血病还会导致视力问题、乏力、出血、骨骼脆弱和神经系统问题。
医生建议我们通过摄入水果和蔬菜来为身体补充更多维生素 C。但有时这可能不足以补充体内缺乏的维生素 C。从化学角度来说,坏血病的发生是因为一些重要的双加氧酶失活了。这些酶依赖于抗坏血酸,属于 2-酮戊二酸依赖性双加氧酶(2-ODDs)类。该酶的机制需要 Fe2+、2-酮戊二酸和 ASC 作为辅助底物才能启动反应。总的来说,2-ODDS 会将 O2 转化为有机底物。
每个 2-ODDS 在机制中都有自己的职责。因此,一些 2-ODDS 会催化羟基化,而另一些会进行去饱和和环闭合或扩展。坏血病是一种可以通过抗坏血酸治疗的疾病。科学家通常将这种过程称为 ASC 给药。它的作用是保持 Fe2+ 稳定。肽酰脯氨酰 4-羟化酶(P4H)的机制使用该酶在多肽链内的脯氨酰残基的第 4 个碳原子进行翻译前羟基化。因此,该酶会将氧气分解为有机底物,以便它们可以用于 2-酮戊二酸的脱羧和脯氨酸的氧化。ASC 被用作 Fe2+ 离子机制的另一个受体,因此它可以在不需要羟基化的情况下驱动机制。由于铁离子的机制非常复杂和反应性,ASC 和 2-ODDS 可能需要经历分子共同进化才能进行反应。当体内缺乏 ASC 时,会导致 P4H 失活,最终导致坏血病。因此,胶原蛋白残基不会被羟基化,胶原蛋白三聚体也不会形成。具体来说,需要进行脯氨酸的羟基化来确保胶原蛋白的折叠。结果,胶原蛋白的折叠会形成三螺旋结构,然后形成有助于热稳定性的纤维。这些胶原蛋白折叠有助于维持皮肤、肌腱、软骨、骨骼、牙齿、角膜、肌肉和血管。因此,这个过程非常重要。科学家已经发现了 ASC 缺乏与胶原蛋白无法折叠之间的关系。例如,他们意识到,自由 ASC 饮食的豚鼠体内 IV 型胶原蛋白含量较低,导致羟基化脯氨酸含量较低。结果,豚鼠患上坏血病,导致血管缺陷。由于胶原蛋白消耗大量的 ASC,而 ASC 缺乏会导致坏血病,因此我们可以得出结论:胶原蛋白的无法折叠在坏血病中起着重要作用。
另一组依赖于 ASC 的双加氧酶可以驱动 DNA 序列中甲基化碱基的修复。其他依赖于 ASC 的植物双加氧酶可以帮助合成信号分子的激素,例如赤霉素和乙烯。
ASC 在 HIF 羟基化信号传导中起着重要作用。HIF1 被称为 HIF 成员之一,它可以激活数百个与营养物质转运、细胞迁移、血管生成和能量代谢相关的基因。HIF 1 具有 2 个亚基:α 和 β 亚基。科学家已经发现,由双加氧酶催化的机制需要包括氧气。驱动氧气机制的是 2 个脯氨酰残基的羟基化。人类中有两个脯氨酰残基。它们是 HIF-α 的 Pro402 和 Pro564。它们被 3 种不同的羟化酶(HIF-P4H)羟基化,并且它们执行与胶原蛋白羟化酶相同的过程。然而,胶原蛋白羟化酶和 HIF-P4H 之间存在差异。HIF-P4H 可以存在于细胞质中,而胶原蛋白羟化酶存在于内质网中。在 HIF-P4H 中,该酶对 O2 底物的亲和力或 O2 的 Km 值高于大气浓度,这意味着它们可以在机制中识别氧气。当氧气可用时,我们称之为常氧条件。在这种情况下,2 个脯氨酰残基将被羟基化。然后导致多蛋白复合物的结合,以攻击蛋白 pVHL 并降解 HIFα。氧气浓度较低的机制称为缺氧,而没有氧气可用的情况称为无氧。当这些条件发生时,羟基化无法进行。结果,HIFα 不会与蛋白 pVHL 结合,导致 HIFα 和 HIFβ 在细胞核中结合。氧气的存在可用于激活机制中的羟基化,并且它的存在可以使 ASC 在反应中变得不那么重要。科学家已经发现,在常氧和缺氧条件下,ASC 起着重要作用。ASC 有助于降低 HIF1 蛋白含量,同时增加 HIFα 降解速率。当向反应中添加 ASC 时,它可能有助于增加缺陷细胞中的 HIF 羟基化。另一项研究表明,降低 ASC 浓度会导致 HIFα 羟基化速率降低,这可能导致这种蛋白降解速率降低。总的来说,ASC 发挥着多种作用,例如它可以通过降低 Fe3+ 的含量来激活酶,并且它可以作为底物与酶结合。
科学家已经对 ASC 在癌症治疗中的作用进行了不同的实验。他们发现,增加 ASC 的浓度会导致 HIF 下调。由于 HIF1α 在癌症治疗中表达,因此有实验表明,癌症患者体内缺乏 ASC 表达。科学家一直在研究 ASC 的功能如何帮助患者体内的免疫细胞。
ASC 在基因表达中发挥了多种作用。研究表明,维生素 C 可以帮助基因转录过程,从而稳定 mRNA。例如,酪氨酸羟化酶的转录是由维生素 C 催化的。此外,ASC 可以通过产生更多胶原蛋白来分化间充质细胞的成员。间充质细胞的成员包括软骨细胞、心肌细胞和成骨细胞。在成骨细胞中,由成骨细胞产生的结合蛋白骨钙素可以由 ASC 表达。当 ASC 增加导致骨钙素基因转录时,就会发生该过程。此外,在腓骨肌萎缩症的实验中使用了小鼠模型。ASC 有助于小鼠髓鞘形成,从而找到了治疗该综合征的新途径。
Tullio, Mario C. De. Beyond the antioxidant: The double life of Vitamin C. 12/6/12
Lantipeptides 以前被称为 lantibiotics,因为它们表现出抗菌特性,但自从发现没有执行抗菌任务的 lantibiotics 后,该术语已更改为 lantipeptides。随着这一发现,它们所属的家族扩展到 90 多种化合物。
Lantipeptides 是核糖体合成的肽,具有硫醚交联,该交联是通过丝氨酸/苏氨酸残基脱水以及随后将半胱氨酸残基添加到生成的脱氢氨基酸 meso-lanthionine (Lan) 和 (2S,3S,6R)-3-methyllanthionine (MeLan 而形成的。Meso-lanthionine 和 (2S,3S,6R)-3-methyllanthionine 是来自前体肽翻译后修饰的交联。
Lantipeptides 以前被称为 lantibiotics,因为它们表现出抗菌特性,但自从发现没有执行抗菌任务的 lantibiotics 后,该术语已更改为 lantipeptides。随着这一发现,它们所属的家族扩展到 90 多种化合物。
Lantibiotics 历史上一直被用作抗菌剂。一种 latipeptide (nisin) 在食品行业中被用作防腐剂已有 50 多年了。Lantibiotics 这个名称最初是作为“含羊毛硫氨酸的肽类抗生素”一词的缩写而引入的 [1]。Erhard Gross 和 John L. Morell 在 1960-70 年代进行的工作导致了当今的 lantibiotics 领域。

Nisin 是一种广为人知且被广泛研究的 lantibiotic。Nisin 由 34 个氨基酸组成,已被广泛用于奶酪、肉类和其他日常用品等产品中,以抵御可能腐败或感染食物的细菌。
最近,对 nisin 进行的研究表明它可能有助于对抗癌症。在密歇根大学进行的一项研究中发现,nisin 在癌细胞的细胞膜上形成孔。这些孔允许钙流入癌细胞,最终会导致其死亡。目前尚不清楚钙流入如何具体导致死亡,但一种由 nisin 激活的蛋白质 CHAC1 参与其中。还发现 nisin 可能中断癌细胞的细胞周期,同时不影响正常细胞 [2]。

lantibiotics 最显着的特性之一是它们对细菌的高活性。它们对革兰氏阳性菌(如葡萄球菌、链球菌、肠球菌和梭状芽孢杆菌)和革兰氏阴性菌(如奈瑟菌)有效。由于这些特性,它们一直是许多科学研究的主题,并在食品保存等行业中得到应用。它们已被发现可治疗各种感染,如艰难梭菌感染等 [3]。Lantipeptides 也可能在其他行业中得到应用,如农业、兽医学和分子成像。
lantipeptides 获得抗菌特性的机制通常是通过抑制细菌细胞壁合成。它们还已知在膜中形成孔,这会极大地破坏细胞的完整性。具体来说,lantipeptides 已知的作用机制是通过抑制转糖基化。它们通过与脂类 II 结合来做到这一点,脂类 II 是构建细胞膜的不可或缺的一部分 [3]。
nisin 能够很好地发挥抗菌作用的机制已被充分记录,这也是它今天被广泛使用的主要原因之一。它通过 A 环和 B 环与焦磷酸基团接触与脂类 II 结合。然后它通过插入自身,形成四分子脂类 II 和八个 nisin 肽的基团,在膜中形成孔 [4]。
Lantipeptides 可以分为四类不同的生物合成酶,这些酶识别 Lan 和 MeLan:脱水酶和环化酶(I 类)、双功能羊毛硫氨酸合成酶(II 类)、三功能合成酶和碳环(III 类)、三功能羊毛硫氨酸合成酶(IV 类)。
I 类 lantipeptides 被称为专门的脱水酶和环化酶,原因很充分。有两种不同的酶执行这些过程:脱水酶 LanB 和环化酶 LanC。LanB 基因提供约 1000 个残基的蛋白质,这些蛋白质与任何已知酶没有同源性。LanC 基因编码约 400 个残基的蛋白质,并呈现出较低的序列同一性。在最近发现和分离的 lantipeptides 中,有一种来自放线菌 Microbispora corallina 的修饰肽,对革兰氏阳性菌具有高活性,以及源自放线菌 Planomonospora 的 planosporicin。这两种肽的 NMR 结构已显示出构象相似性。 [3]
II 类 lantipeptides 被称为双功能羊毛硫氨酸合成酶。已知它们执行脱水和环化反应。LanM 是执行这些过程的双功能合成酶。它产生的蛋白质长度在 900-1,2000 个残基之间,包含两个结构域,即一个 N 端脱水酶结构域,它没有与 LanB 同源的酶,以及一个 C 端环化酶结构域,它与 LanC 具有四分之一的序列同一性,包括对 NISC 催化至关重要的锌结合残基的保守性。最近,该类 lantipeptide 有许多新成员加入。其中之一是 haloduracin,它来自 Bacillus halodurans。这是第一个来自嗜碱种类的 lantipeptide,这意味着它可以在高 pH(或碱性)环境中生存。 [3]
III 类 lantipeptides 被称为三功能合成酶和碳环。生物合成存在于来自链霉菌 Streptomyces coelicolor 的形态发生肽 SapB 中。II 类和 III 类 lantipeptides 之间存在很大差异,导致了单独的命名。差异在于这种肽没有表现出抗生素活性。相反,它促进与链霉菌孢子形成相关的营养菌丝的生长。基因簇中包含一种名为 RamC 的假定的修饰酶。这种酶类似于丝氨酸/苏氨酸蛋白激酶,并以 C 端结构域构建,该结构域具有与 LanM 的环化酶结构域相似的同源特征。然而,锌结合缺失 [3]。
2010 年,从放线菌 Actinomadura namibiensis 中发现了迷宫肽。发现被称为 LabKC 的三功能 lantipeptide 与以前已知的 RamC 具有同源性。发现迷宫肽是 LabKC 的修饰产物之一,它有助于缓解小鼠的神经性疼痛。这是以前未观察到的 lantipeptides 的功能 [3]。
IV 类 lantipeptides 生物合成涉及链霉菌 Streptomyces venezuelae 中的隐性基因簇。合成酶 VenL 由一个 N 端 OspF 样裂解酶结构域组成,该结构域具有一个丝氨酸/苏氨酸激酶结构域中心。与 RamC 不同,VenL 的 C 端环化酶结构域包含存在于 LanC 和 LanM 中的锌结合基序 [3]。
了解Lantipeptides可以帮助以有利且可适应的策略,以低遗传成本产生大量的化学多样性。Lantipeptide生物合成一直通过发现新的生物合成机制类别、新的翻译后修饰以及更多编码Lantipeptide的基因簇而发展。尽管一些催化机制仍然未知,例如LanB,但是,通过发现大肠杆菌中生产的方法,有助于LanB的调查,此外,通过终止密码子抑制技术将非蛋白氨基酸引入Lantipeptides。
Lantipeptide工程的一种方法是在大肠杆菌中进行。这在2005年首次进行,当时生产了截短的nukacin ISK-1。这是通过在一个载体上共表达nukA和nukM来完成的。在2011年,生产了修饰的LanAs。这是通过共表达lanA和lanM来完成的。在一个载体上共表达nisA和nisB,在另一个载体上共表达nisC,会导致生产nisin前体肽。这是唯一一个在文献中有记录的大肠杆菌中产生的I类Lantipeptide[3]。
体外工程方法存在自身的一系列问题,主要原因是免疫或输出方面的问题。这种方法利用合成底物。这种合成方法的一个缺点是蛋白水解,它通常会导致低产量。为了提高这种体外方法,需要优化这一步[3]。
- ↑ Heike, Brotz和Hans-Georg Sahl "Lantibiotics作用机制的新见解——通过结合相同的分子靶点产生不同的生物学效应", [Oxford Journals], 2012. 于2012年12月7日检索。
- ↑ "常见的食品防腐剂可以减缓甚至阻止肿瘤生长", MedicalPress, 2012. 于2012年12月7日检索。
- ↑ a b c d e f g h i "Lantipeptides的发现、生物合成和工程". 2012.
{{cite web}}: Unknown parameter|retrieved=ignored (|access-date=suggested) (help) - ↑ Shang-Te D Hsu 等人 "Nisin-脂类II复合物揭示了为新型抗生素提供蓝图的焦磷酸盐笼", [Nature.com], 2004. 于2012年12月7日检索。
ADME是药理学和药代动力学中常用的缩略语,用于指代药物/药品在人体内的四个基本阶段:吸收、分布、代谢和排泄。
吸收是指药物从其剂型释放后进入血液的过程。人体可以通过多种方式吸收药物,例如口服(吞服泰诺片剂)、肌内注射(在手臂肌肉中注射流感疫苗)、皮下注射(在皮肤下方注射胰岛素)、静脉注射(通过静脉接受化疗)或透皮吸收(使用皮肤贴剂)。最常见的方式是口服给药。一旦药物被吸收,它就会被特殊的血管输送,从消化道进入肝脏,在肝脏中,大量的药物可能被代谢酶破坏(通常称为“首过效应”。影响口服药物吸收的最重要因素之一是胃排空时间。胃排空时间是指药物在胃中停留的时间,然后排空进入小肠。这个时间会影响药物的作用,因为大多数药物是在肠道中吸收的。胃酸会在药物到达吸收阶段之前破坏许多药物。一旦药物离开胃而不被破坏,它在肠道中的移动速度会影响其目前的吸收。如果药物在肠道中缓慢移动,更多的药物会被吸收,因为它与肠道膜接触的时间更长。另一方面,快速通过肠道可能会导致药物未被完全吸收。此外,胆汁盐和肠道酶也会影响药物的吸收。胆汁盐会改善一些疏水性药物的吸收。然而,酶会通过破坏药物来降低药物的吸收,因为药物会通过肠道。
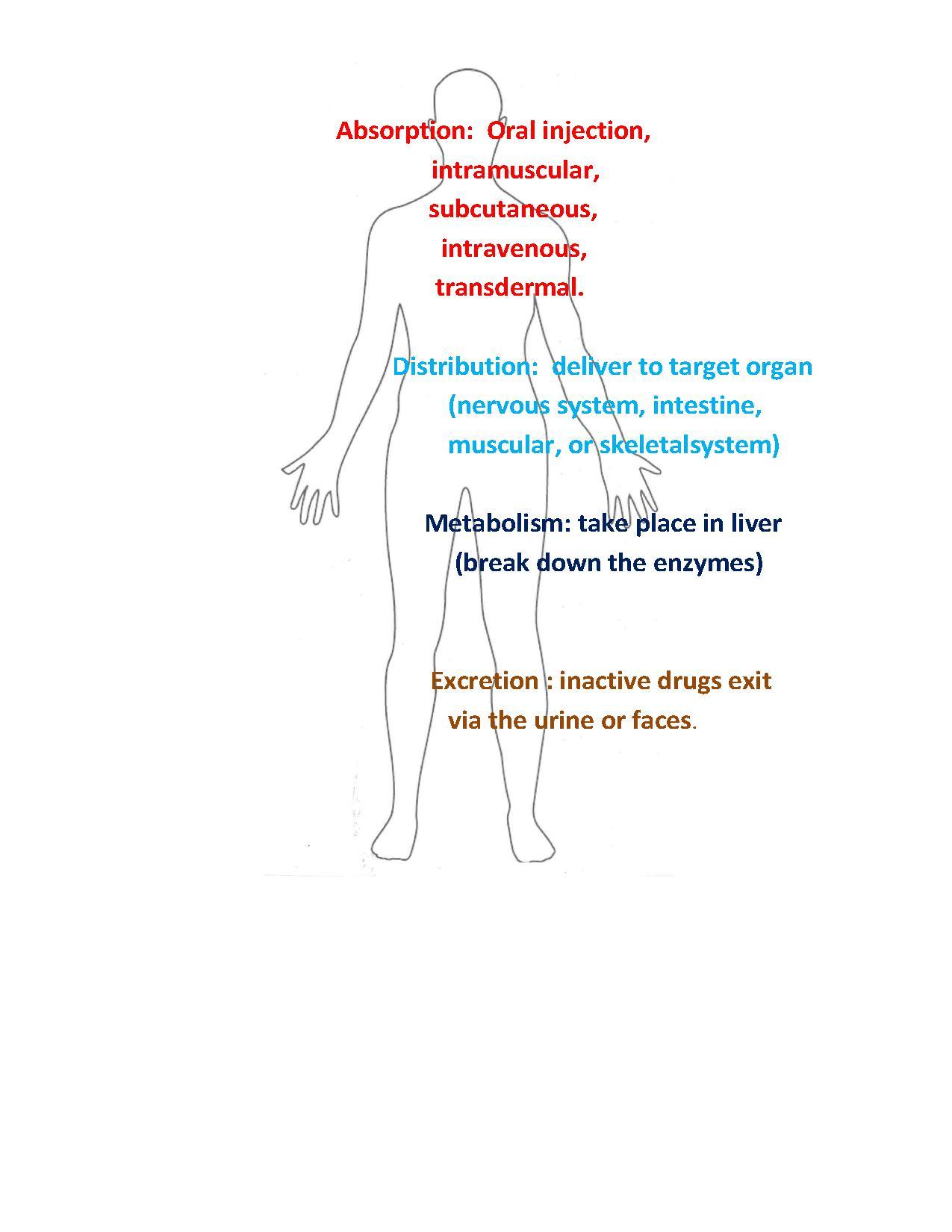
分布是指药物进入血液后在体内的移动。血液将药物输送到全身,也输送到其作用部位。首先,分布受某些器官血流速度的影响。当一个器官的血流速度很高,比如心脏、肝脏和肾脏时,药物会很快分布。然而,血流速度慢的器官,比如肌肉、脂肪和皮肤,会导致分布速度慢得多。此外,组织膜对药物的通透性也极其重要。小的药物分子和疏水性药物很容易扩散通过组织膜。此外,还有一些组织膜具有专门的转运机制,有助于渗透。然而,也有一些组织膜对允许药物穿透具有高度选择性。例如,血脑屏障限制了药物进入大脑。最后,蛋白质结合也会影响分布。大多数药物与血浆中的蛋白质结合,形成所谓的复合物。由于这些复合物很大,它会阻止药物进入其作用部位。因此,只有游离的或未结合的药物才能通过组织膜移动。此外,一些结合能力更强的药物可以将结合能力弱的药物从蛋白质上置换下来,导致结合能力弱的药物不再结合。
药物代谢是指人体处理药物的方式。这种在体内被转化的药物被称为代谢物。大多数代谢物是不活跃的分子,会被排泄,但有些是活跃的,会在人体内产生影响,直到它们被进一步代谢或排泄。肝脏是药物代谢的主要部位。肝脏内的酶与药物相互作用,并将它们转化为代谢物。CYP3A4是一种肠道中的药物代谢酶,它会增加或改变某些药物在人体内的血药浓度。CYP450是一种细胞色素酶,它处理必需的分子,如激素和维生素,以及代谢数百种处方药和天然物质。一些药物会使肝脏增加其酶活性,导致药物代谢更快。因此,一些药物的剂量必须更大才能产生相同的治疗效果。然而,一些药物会降低酶活性。在这种情况下,需要更小的药物剂量以避免毒性。此外,肝脏可能会将药物分泌到胆汁中,胆汁储存在胆囊中。胆汁中的任何药物或代谢物可能会被重新吸收或随粪便排出。如果药物被重新吸收回血液,则称为肠肝循环。
大多数药物及其代谢产物通过肾脏排泄,并通过尿液排出体外。一些药物不易被胃肠道吸收,因此会通过粪便而不是尿液排泄。排泄也可以通过胆汁进行,一些药物通过肺部呼出的气体排出体外。但是,大多数药物都是通过肾脏排泄的,肾脏会过滤血液并清除血液中的废物。一部分血浆水通过称为肾小球滤过的过程从血液中过滤到肾小管。这种过滤的水中可能包含来自身体其他部位的废弃药物。随着水继续在体内流动,其他废弃药物会被分泌到液体中。与此同时,一些药物可以通过尿液重吸收回到血液中。在所有这些过程完成之后,最终的液体以尿液的形式从体内排出。尿液排泄的速度远快于粪便排泄的速度。通过尿液排泄的药物需要几个小时,而通过粪便排泄的药物则需要几天。
- 药剂技术员. 纽约: 莫顿出版公司, 2007.
- 戴维斯, 艾莉森. 药物设计. [贝塞斯达, 马里兰]: 美国卫生与公众服务部, 国家卫生研究院, 国家普通医学科学研究所, 2006. 印刷版.